0
留言稍后联系!

发布时间:2019-11-02所属分类:医学论文浏览:1490次
摘 要: 摘要 乳腺癌是影响女性健康最主要的恶性肿瘤之一.表观遗传修饰及活性氧(ROS)过度积累引起的氧化应激在乳腺癌发生发展中起关键作用,表观遗传修饰与 ROS 的生成和清除相互影响.本文通过对目前有关表观遗传修饰和 ROS 参与乳腺癌的发生发展进行综述,为寻求乳

摘要 乳腺癌是影响女性健康最主要的恶性肿瘤之一.表观遗传修饰及活性氧(ROS)过度积累引起的氧化应激在乳腺癌发生发展中起关键作用,表观遗传修饰与 ROS 的生成和清除相互影响.本文通过对目前有关表观遗传修饰和 ROS 参与乳腺癌的发生发展进行综述,为寻求乳腺癌发生发展的生物标志物及精准治疗提供思路。
关键词 表观遗传修饰,活性氧,氧化还原状态,乳腺癌
Globocan 肿瘤流行病学数据显示,2012 年全球女性乳腺癌新发病例与死亡数位居女性恶性肿瘤首位,中国近年女性乳腺癌发病例数和死亡例数在世界范围位居前列,其发病增长速度超全球 2 倍,成为女性发病率最高的癌症.乳腺癌的异质性和在病因学上相关的基因组演变,使现有治疗方案总体效果不尽人意,所以寻找新的作用靶点将对乳腺癌的治疗研究具有重要意义.表观遗传机制已经成为乳腺癌发生进展的基本参与者,因其变化的可逆性可作为治疗乳腺癌的靶标.研究表明乳腺癌组织具有较强的氧化应激状态和明显的抗氧化能力[1].表观遗传修饰与 ROS 的相互作用影响着乳腺癌的发生发展,研究表观遗传修饰与 ROS 的相互作用可能对研究乳腺癌中的靶向治疗提供新途径,为精准治疗及个性化治疗提供可能的机会.
1 乳腺癌中的表观遗传修饰
表观遗传是指核苷酸序列不发生变化的情况下,基因的表达活性发生了可遗传的变化,包括 DNA 甲基化、组蛋白修饰、非编码 RNA 调控及染色体重塑等,会导致基因转录失调和细胞增殖失常,从而致癌.其中以 DNA 甲基化和组蛋白修饰的研究最为重要.表观遗传修饰因具有可逆性变化的特点,可以作为治疗乳腺癌的靶点 . Teschendorff 等[2]证明在邻近癌症的正常组织中存在数以万计的表观遗传学改变,从而支持乳腺癌在表观遗传领域存在修饰.癌发生早期阶段,DNA 甲基化和组蛋白修饰引起的染色质结构改变影响细胞可塑性,促进乳腺细胞获得不受控制的自我更新特性;癌发展后期阶段,添加的表观遗传学改变及微环境的信号会调节癌细胞表型并影响肿瘤的转移倾向[3].
1.1 DNA 甲基化
DNA 甲基化指 DNA 复制后,S 腺苷甲硫氨酸 (SAM)上的甲基基团在 DNA 甲基转移酶(Dnmt)催化下连接到 DNA 分子胞嘧啶上形成 5- 胞嘧啶的过程,主要位点是富含 CpG 二核苷酸的 CpG 岛. CpG 岛常位于转录调控区附近,多项研究证实基因组整体低甲基化往往伴随着局部的 DNA 高甲基化及该区域基因表达沉默.肿瘤发生早、晚期 Dnmt 表达增加,表明 DNA 异常甲基化诱导了肿瘤的发生和发展.Leu 等[4]利用 RNA 干扰技术发现,在癌细胞中所有 Dnmt 都协同性地过度表达(尤其是 Dnmt1 和 Dnmt3b)以维持 DNA 甲基化和基因沉默.原癌基因 KIT 受体酪氨酸激酶在调节细胞增殖、存活和迁移过程中发挥重要作用. Radoslav 等[5]的研究表明,与正常乳腺组织相比, KIT 启动子在乳腺肿瘤中高甲基化.Good 等[6]通过乳腺癌的生物信息学分析,发现 TET1 的低甲基化会激活致癌途径,这种低甲基化与激活 PI3K 突变相互排斥,表明去甲基化可能是激活该致癌途径的替代机制.在乳腺癌中,已有 100 多个基因的启动子发生甲基化,且这些基因在 DNA 修 复 ( 如 BRCA1)、组织侵入转移和调节细胞转录中发挥重要作用.研究乳腺癌的甲基化差异,可能针对个体的不同甲基化修饰提出个性化治疗方案,这符合精准医学的要求.
1.2 组蛋白修饰
组蛋白是核蛋白染色质中与 DNA 分子密切相关的结构,在基因表达调控中发挥重要作用,共有 5 种类型:H1、H2A、H2B、H3 和 H4.其中 H1 作为连接蛋白参与染色质的高级结构,其他 4 种与 DNA 结合形成核小体.翻译后修饰是表观遗传调控的一个主要组成部分,根据细胞核内的信号环境,破坏 DNA 与组蛋白的相互作用改变核小体结构,或形成转录调节因子的结合位点来影响染色质结构,最终影响病理细胞反应.肿瘤细胞的主要特征之一是组蛋白高甲基化和低乙酰化.2014 年李海涛教授课题组[7]的研究首次揭示出生物体内存在组蛋白变体特异的甲基化识别蛋白.这种对组蛋白变体和甲基化修饰类型的双重识别,体现了真核生物表观遗传调控的复杂性和重要性,相关结构研究成果也为基于肿瘤抑制因子 ZMYND11 靶向的小分子抑制剂筛选提供了重要理论基础.
1.2.1 组蛋白甲基化
大部分组蛋白甲基化酶(KMT)含 SET 结构域,使组蛋白发生 2 倍或 3 倍甲基化,导致染色体空间结构松散,影响下游转录因子与靶基因的结合,最终促进肿瘤细胞生长、侵袭和迁移.组蛋白去甲基化酶(KDM)分两类:一类为赖氨酸特异性组蛋白去甲基化酶 1(LSD1),对调控上皮 - 间质细胞转换 (EMT)的基因起重要作用;另一类是含 JmjC 结构域的蛋白质家族,可对组蛋白多个位点进行去甲基化,调控转录、细胞增殖和肿瘤发生等多种生物学过程.
组蛋白甲基化引起的基因表达、激活或抑制取决于被修饰的氨基酸残基.如 组 蛋 白 H3 上 赖氨 酸 4 的单甲基化或三甲基化 (H3K4me1 或 H3K4me3)和赖氨酸 36 上的三甲基化(H3K36me3) 与开放染色质激活转录有关,而组蛋白 H3 上的赖氨酸 9 和 27 的三甲基化(H3K9me3 和 H3K27me3) 与致密染色质抑制基因表达有关[3].LSD1 在雌激素受体阴性乳腺癌中高表达,与癌细胞的侵袭性有关,LSD1 减少会导致体外生长抑制[8].Messier 等 [9] 使 用 全 基 因 组 ChIP-Seq 方 法 评 估 正 常 (MCF-10A)、 致 瘤 性 (MCF-7)、 促 转 移 性 (MDA-MB-231) 3 种乳腺癌细胞系,发现启动子处 H3K4me3 的增加主要发生在三阴性 MDA-MB-231 细胞系中,表明组蛋白甲基化可能与增加乳腺癌的转移潜能相关.Al Emran A 等[10]使用乳腺癌细胞系生成诱导药物耐受细胞(IDTC),在 IDTC 中观察到 H3K4me3 和 H3K27me3 的缺失和 H3K9me3 标记增加,这是对药物暴露或营养饥饿的明显反应,而且这些表观遗传变化在停止用药后可逆.
1.2.2 组蛋白乙酰化
乙酰化由组蛋白乙酰基转移酶(HAT)和去乙酰化酶(HDAC)共同调控,是一种可逆的蛋白质共价修饰形式.组蛋白的乙酰化可以减少赖氨酸残基与 DNA 的相互作用,使组蛋白与 DNA 分离,利于打开染色质使其成为开放结构,促进转录因子与 DNA 结合,启动基因的转录和表达.研究证实, HAT 有抑制肿瘤的功能,但 HAT 的过量表达同样可以导致癌症的发生[3],当 HDAC 过量或 HAT 的数量减少时,组蛋白乙酰化的平衡将偏向去乙酰化,从而导致基因表达的调节异常,因此,HAT 和 HDAC 之间的失衡引起的表观遗传变化会严重影响基因的转录且与肿瘤的发生和进展相关.由此可见,肿瘤细胞内过表达的 HDAC 可以作为精准治疗的靶蛋白.
组蛋白去乙酰化酶抑制剂(HDACi)可竞争性抑制底物蛋白质与 HDAC 结合,促进组蛋白乙酰化修饰,利于转录.Muller 等[11]未发现 HDAC1 或 HDAC2 与乳腺癌的预后有任何关联,而 Schech 等[12] 证明 HDACi 能有效地选择性抑制Ⅰ类 和Ⅳ类 HDAC,且能够逆转乳腺癌细胞系中的 EMT. Messier 等[9]证明组蛋白 H3 的赖氨酸 4 的乙酰化 (H3K4ac)标记增加是乳腺癌恶性转化所必需的,组蛋白 H3 的赖氨酸 9 的乙酰化(H3K9Ac)与开放染色质区域相关.Cui Z 等[13]的研究表明,乳腺肿瘤中更常见到 HDAC3 的高表达、HDAC3 高表达与乳腺肿瘤的 ER 阴性表达、PR 阴性表达、HER2 过表达、PT 分期和临床分期密切相关,并得出 HDAC3 可能是浸润性导管乳腺癌的一个合适的预后指标.总体而言,这些数据表明特定组蛋白修饰在改变乳腺癌表观基因组中起关键作用,并导致乳腺肿瘤的异质性.因此,我们可以通过靶向特定组蛋白修饰位点为乳腺癌提供治疗方案.
2 氧化还原状态与乳腺癌
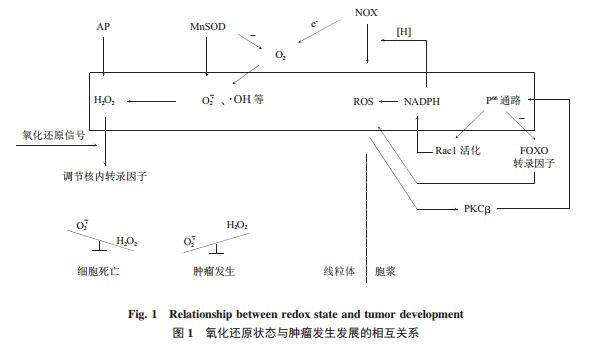
ROS 又称“氧自由基”,主要产生于线粒体,是需氧生物体内氧分子经呼吸链中的单电子逐步还原形成的一类物质,具有很强的氧化性,包括过氧化 氢(H2O2)、超氧 阴 离 子(O2 ·)、羟自 由 基(·OH) 等.胞内的 ROS 大部分来自于线粒体的氧化磷酸化过程,还有部分来自胞浆,如 NADPH 氧化酶和 PKCβ/p66Shc 通路,它们可直接感受外源刺激,诱导胞内产生 ROS.同时机体自身也有一套完整的抗氧化防御系统,使细胞内 ROS 的产生和降解处于平衡状态,保护细胞膜和组织免受损害.抗氧化系统包括两部分:第一部分为抗氧化酶类,包括超氧化物歧化酶(SOD)、过氧化氢酶(CAT)和谷胱甘肽过氧化物酶(GSH-Px)等;第二类是含巯基的小分子蛋白质,包括硫氧还蛋白(Trx)和还原性谷胱甘肽(GSH)等.当机体遭受有害刺激使细胞内 ROS 不能被及时有效清除而过度积累时,氧化系统失衡,出现“氧化应激态”,引起机体多种分子(线粒体、DNA、蛋白质等)的氧化损伤.
乳腺癌的转移能力与细胞氧化还原状态密切相关.氧化还原失衡可以通过氧化还原依赖性的活化蛋白激酶 C(PKC)促进生长因子和整合素信号传导,导致丝裂原活化蛋白激酶(MAPK)信号级联和肿瘤细胞迁移[14].ROS 在癌症中有双重用途,它不仅可以激活细胞凋亡途径杀死癌症细胞,还可保护正常组织免受放射、化疗毒性.ROS 存在这种潜力是因为恶性细胞的增殖和自我更新依赖细胞内 ROS 的升高,且基于正常健康细胞不具有升高 ROS 水平的可能[15].氧化还原状态与肿瘤发生发展的相互关系如图 1 所示.
2.1 H2O2 水平与乳腺癌
H2O2 是 O2 的双电子还原产物,目前至少已有 30 种酶被鉴定为 H2O2 产生酶[16],包括超氧化物歧化酶(SOD)、NOX 和黄素蛋白脱氢酶.H2O2 可作为信使携带一个产生的氧化还原信号到达目标部位,调节转录因子 AP-1、Nrf2、CREB、HIF-1α、 p53、NF-κB、SP1 的活性和 EMT[17].H2O2 浓度依赖于 AP,同时对维持稳态有关键作用.H2O2 水平可能对细胞生长有正面或负面的影响,这取决于 H2O2 增加的水平及所在细胞位置.已经表明,O2 · 与 H2O2 的比例决定了癌症的走向:O2 ·过多支持细胞存活并促进肿瘤发生,而 H2O2 过多则诱导细胞死亡信号传导[18].乳腺癌细胞在 DNA 损伤程度很高时,在低浓度 H2O2 刺激下进行细胞分裂,而较高浓度会抑制细胞生长并最终导致细胞死亡[15].
2.2 锰超氧化物歧化酶 (MnSOD) 与乳腺癌
SOD 是锰依赖性酶,可抑制信号传导 . MnSOD 是控制线粒体中 ROS 量的关键因素,至少有 2 个主要功能:一个是抗氧化功能,防止过量 O2 导致 O2 ·积累的防御机制;另一个功能是能将 O2 ·转化为 H2O2.相比于正常细胞,侵略性癌症中的 MnSOD 和氧化应激增加.MnSOD 具有肿瘤抑制和促进功能,在癌症中的双重作用主要与其在癌症的某个阶段是作为线粒体 ROS 清除剂还是 H2O2 生成剂有关[19].如果 MnSOD 是 H2O2 生成剂,则 MnSOD 的过表达会抑制癌症生长,但如果 MnSOD 是一种线粒体超氧化物清除剂,MnSOD 的抑制将增强癌细胞死亡.研究结果支持 MnSOD 在抵抗 ROS 介导的损伤,保护人正常组织中的作用[15].人乳腺上皮细胞 MCF-10A 中的 MnSOD 活性在增殖阶段比静止状态明显下降[20],乳腺癌细胞中表现出显著的 MnSOD 上调,其通过影响细胞氧化还原环境来调节 EMT 和 MET 相关表型之间的转换.另一方面,MnSOD 过度表达显著降低乳腺癌细胞的存活和生长转移[21].
2.3 胞浆内 NADPH 氧化酶及 PKCβ/p66Shc 通路与乳腺癌
NADPH 氧化酶的催化亚基 gp91phox 及其同系 物 NOX1、 NOX3、 NOX4、 NOX5、 DUOX1、 DUOX2 被称为 NOX 家族.当细胞接收到外源刺激时,异常激活的 NOX 家 族 蛋 白 以 NADPH/ NADH 为递氢体进行催化反应,迅速生成高浓度的 ROS 参与诱导肿瘤的发生和发展.在这些酶催化的反应中,电子受体是氧,电子转移反应的产物是 O2 ·,由于自发和酶促歧化,H2O2 也迅速产生. NOX1 在乳腺癌中过表达,与生长、增殖和侵袭有关;NOX4 在乳腺癌中的过表达与迁移、血管生成、糖酵解转变和预后不良有关[22].Shen 等[23]的研究表明使用天然酶抑制 NOX1,降低了 ROS 的产生并在乳腺癌中减少了癌症的转移.总体而言, NOX 酶的活化或增加 NOX 表达,可以作为乳腺癌的标记物以指示其对给定的 ROS 生成的敏感性或以酶为基础的抗性治疗.
外源刺激导致胞内 ROS 升高的主要原因之一是 p66Shc 的活化.p66Shc 是 ShcA 基因编码的蛋白质,可感受外源刺激及胞内 ROS 的水平,从 3 个方面诱导 ROS 的生成:a.在细胞核中,p66Shc 抑制 FOXO 转录因子的活性,导致 ROS 清除酶的表达 降 低 , 减 少 ROS 的 清 除 ;b.在 细 胞 浆 内 , p66Shc 可促进 racl 分子活化,诱导 NADPH 氧化酶产生 ROS,此外,p66Shc 也存在于线粒体膜间隙; c.积聚的 ROS 通过氧化应激激活 PKCβ,引起 p66Shc 磷酸化而被活化以及线粒体转位,促进线粒体产生并释放 H2O2,从而进一步提高细胞内 H2O2 的水平,触发线粒体促凋亡作用.胞浆内 ROS 增加,促进 PKCβ/p66Shc 自我激活回路,维持或增加 PKCβ 的激活,从而不断诱导胞浆中 ROS 产生[24].
3 ROS 引起的氧化应激与表观遗传修饰的交互影响在乳腺癌中的作用
ROS 引起的氧化应激会导致基因表观遗传变化,与肿瘤发生密不可分.外源刺激长期反复作用导致氧化系统失衡,导致机体长期处于过量 ROS 水平,以致引发癌症.表观遗传修饰可以通过改变抗氧化酶在胞内的表达水平,影响 ROS 的清除效率;同时抗氧化酶的表观遗传修饰可以诱导 ROS 产生,该过程是一个 ROS 通过引起表观遗传修饰诱导抗氧化酶表达沉默,而减少自身清除的正反馈过程[25].机体的酶促反应是瞬时性的,而表观遗传修饰可以长期稳定地发挥作用,被固定下来,提供适应疾病发生的分子环境.ROS 引起的氧化应激与表观遗传修饰的交互影响,在乳腺癌中的作用机制如图 2 所示.
3.1 ROS 引起的氧化应激与 DNA 甲基化
DNA 甲基化是氧化应激诱发的一个重要的表观遗传变化,主要表现在高甲基化引起的基因转录抑制(如抑癌基因)和低甲基化诱导基因转录激活(如癌基因),在肿瘤发生发展中起重要作用.氧化应激会引起 CpG 岛 DNA 甲基化水平降低,导致肿瘤发生[26].
ROS(特别是·OH)过量引起的氧化应激导致 DNA 损伤,阻碍了 DNA 作为甲基转移酶的底物,也降低了 DNA 碱基对甲基的接受能力.Veeck 等[27] 证明 DNA 甲基化的异常对于正常乳腺细胞的恶性转化起重要作用.在氧化应激引起的肿瘤中,肿瘤抑制基因是最为常见的甲基化异常位点,如在乳腺癌中肿瘤抑制基因 RB、p16INK4A、p53、BRCA1 等启动子区域 CpG 位点均可发生高甲基化,从而使基因表达沉默[28].p53 突变的肿瘤细胞增殖与凋亡失去控制,导致无限繁殖;BRCA1 等的甲基化水平已作为乳腺癌发生的诊断工具.
3.2 ROS 引起的氧化应激与乙酰化
研究表明,表观遗传修饰可以调控 NOX 家族蛋白的表达从而影响癌症的发生和发展[29].NOX2 调控机体大多数非吞噬细胞中 ROS 的产生,其表达可能受到组蛋白乙酰化的调控.已有研究表明,在 赖 氨 酸 122 处 的 MnSOD 的乙酰基模拟物 (MnSODK122Q)可增加线粒体 ROS 水平,导致基因组不稳定并且与乳腺癌恶性肿瘤风险相关[15].
HDACi 可提高肿瘤细胞内的 ROS 水平,活化多种信号转导途径,启动 DNA 应激反应,抑制肿瘤细胞增殖并诱导其凋亡以发挥抗癌活性 . HDACi 在杀伤肿瘤细胞过程中 ROS 的调控机制: a.过度积累的 ROS 会破坏线粒体的膜电势,增加线粒体膜通透性,活化 caspase 通路,诱导肿瘤细胞凋亡;b.HDACi 可以上调硫 Trx 结合蛋白 2 (TBP2),TBP2 抑制抗氧化剂清除剂 Trx,从而提高细胞内的 ROS 水平,HDACi 引起的乙酰化修饰使 DNA 更易被 ROS 结合并氧化,引起 DNA 氧化损伤,发挥杀伤肿瘤细胞的作用[30].因此,可根据乳腺癌患者细胞的不同乙酰化修饰靶向设计 HDACi 杀伤癌细胞.
推荐阅读:医疗技术方面论文有哪些征稿期刊
从事医学领域工作的技术人员,无论是研究项目还是晋升职称都会要求发表论文的,而且这一领域比其他行业要求更为严格,为此很多想要发表医疗技术论文的作者,对于期刊的选择了解的并不多,哪类期刊能征收这方面的论文,大家是比较困惑的,为此,小编在这里给大家推荐了几本合适的刊物,有需要的可以直接与期刊天空编辑联系。
3.3 表观遗传修饰调控抗氧化酶系统影响 ROS 的清除
抗氧化酶 SOD、GPx 及 CAT 受表观遗传修饰调控与乳腺癌的发生发展密切相关,清除效率取决于抗氧化物酶在胞内的表达水平,以及胞内 ROS 产生或累积的浓度.肿瘤发生早期阶段,SOD2 发生高甲基化降低其表达量,引起胞内 ROS 增加从而促进细胞增殖;当肿瘤开始侵袭和转移时, SOD2 甲基化水平下降,表达量上调,引起胞内 ROS 水平降低,可能更利于细胞的侵袭转移[31]. Nelson 等[32]已经提出 MnSOD 基因表达变化与人乳腺癌细胞的增殖和迁移有关,即抑制抗氧化酶基因的表达与氧化应激引起的启动子区高甲基化所致的基因沉默具有正相关性.有研究认为,抗氧化酶的表观遗传修饰可以通过 ROS 诱导产生,该过程是一个 ROS 通过表观遗传修饰改变诱导抗氧化酶表达沉默,继而减少自身清除的正反馈过程[15].因此,在肿瘤发生过程中,抗氧化酶的表观遗传修饰改变通过影响胞内 ROS 的清除,在机体氧化平衡系统的调控中起关键作用.
声明:①文献来自知网、维普、万方等检索数据库,说明本文献已经发表见刊,恭喜作者.②如果您是作者且不想本平台展示文献信息,可联系学术顾问予以删除.

SCISSCIAHCI

