0
发表咨询在线!

发布时间:2020-04-29所属分类:医学论文浏览:1304次
摘 要: 摘要分别以1,6-己二胺和对苯二胺为原料,与对映体纯L-缬氨酸发生缩合进而得到手性氨基酚化合物。所合成氨基酚及相应的中间产物经过高分辨质谱、核磁共振氢谱和核磁共振碳谱等进行表征并确认结构。此外,化合物2A和7B-1通过X-ray单晶衍射法确定了结构。 关键

摘要分别以1,6-己二胺和对苯二胺为原料,与对映体纯L-缬氨酸发生缩合进而得到手性氨基酚化合物。所合成氨基酚及相应的中间产物经过高分辨质谱、核磁共振氢谱和核磁共振碳谱等进行表征并确认结构。此外,化合物2A和7B-1通过X-ray单晶衍射法确定了结构。
关键词氨基酚,合成,单晶
1.引言随着药物化学的不断发展,使用催化法实现手性合成已经成为十分普遍的方法。迄今为止,过渡金属催化剂、酶、有机小分子催化剂等被广泛使用[1][2][3],特别是近几年来迅速发展起来的手性有机小分子催化剂,因其具有容易制备、价格低廉、对环境友好、反应条件温和等优势而受到科学家们的青睐,成为研究的热点和前沿[4]。
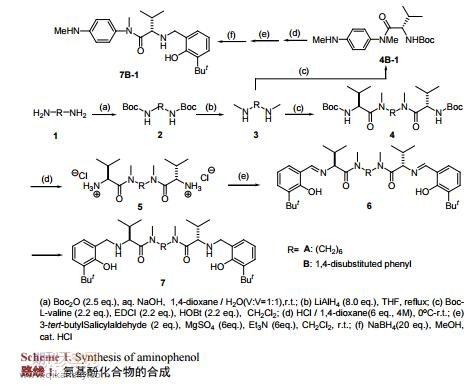
氨基酚化合物,其结构中含有氨基及酚羟基两个官能团,它不仅可以作为配体与金属Ti、Cu等络合后参与反应,也可以单独作为一种双功能有机小分子催化剂使用,其在催化氰基对环氧化合物[5][6]以及亚胺[7][8]的不对称加成、Mannich反应[9]、亚胺的烯丙基化反应[10][11][12]等多种反应中都表现出良好催化活性,相应反应都获得较高的产率和对映选择性。同时,这类催化剂拥有突出的优势:易于合成,方便提纯,催化剂用量少等。因而,合成新颖结构的氨基酚化合物具有重要的意义。本文设计合成了三种手性氨基酚化合物(7A,7B,7B-1),化合物7A和7B分别为含有链式和芳基对称结构的氨基酚化合物,化合物7B-1为含有苯胺结构的氨基酚化合物。氨基酚化合物的合成如路线1所示。
2.实验方法
2.1.主要试剂及仪器
INOVA-600型核磁共振仪(以TMS为内标,CDCl3为溶剂);BruckerEquinox55FT-TR型红外光谱仪(KBr压片);Brucker型质谱仪(ESI电离技术);低温实验使用EYELAPSL-1810型磁力搅拌低温恒温水槽(无水乙醇为介质)。
实验所用试剂均为分析纯,石油醚沸程为60℃~90℃,其中,无水四氢呋喃是在钠砂中惰性气体保护下加热回流并用二苯甲酮显色;柱层析硅胶G(青岛海洋化工有限公司);薄层层析硅胶GF254(青岛海洋化工有限公司);薄层层析板(GF254,加0.5%CMC自制),紫外灯下(254nm)观测并用碘蒸气显色。
2.2.氨基酚化合物的合成
以化合物7A的合成为例[10]:
50mL圆底瓶中加入化合物1A(0.23g,2mmol),添加混合溶剂(20mL,V1,4-二氧六环:V水=1:1)搅拌使其溶解溶液。冰浴条件下分批加入(Boc)2O(1.09g,5mmol,2.5equiv.),同时缓慢滴加10%的NaOH溶液,使PH保持在8~9之间。待酸酐加毕,混合液室温搅拌过夜,TLC监测反应。用乙酸乙酯(20mL×3)萃取,水相用10%的KHSO4溶液酸化至PH=2~3后,用乙酸乙酯萃取水相,合并有机相,无水Na2SO4干燥,过滤,滤液浓缩得到粗产物。粗产物经乙酸乙酯重结晶,得化合物2A。
50mL圆底瓶中加入无水THF,冰浴下加入LiAlH4(0.61g,16mmol,8.0equiv.),缓慢加入2A(0.63g,2mmol),Ar保护下回流4h,TLC监测反应。在冰浴条件下,缓慢加入Na2SO4水溶液淬灭反应,过滤,乙酸乙酯洗涤固体,合并有机相,浓缩得粗产物。减压蒸馏纯化(b.p.70℃,压力44Pa)得无色液体3A。
EDCI(0.17g,0.92mmol,2.2equiv.)、CH2Cl2(20mL)、HOBT(0.12g,0.92mmol,2.2equiv.)、Boc-L-缬氨酸(0.20g,0.92mmol,2.2equiv.)顺序加入50mL圆底瓶中,搅拌溶解。将3A(0.06g,0.42mmol)溶解在CH2Cl2中,逐滴加入到上述混合溶液中,室温搅拌,TLC监测反应。反应完毕,于反应体系中加入10wt%的柠檬酸(20mL),搅拌,将析出的白色沉淀物过滤,滤液用10wt%的柠檬酸(20mL)、饱和NaHCO3、饱和食盐水洗涤,合并有机相,无水Na2SO4干燥,过滤,浓缩得粗产物。快速柱层析(PE:EA=2:1)得无色油状液体4A,直接进行下一步反应。
50mL圆底瓶中加入4A(0.13g,0.24mmol),冰浴搅拌下加入盐酸的1,4-二氧六环溶液(4.0M,2.88mmol,0.72mL,12equiv.),缓慢升至室温搅拌反应,1.5h后往反应体系中充入30min氮气,减压浓缩,得到棕褐色粘稠液体5A,不经纯化直接进行下一步反应。
氩气保护下,在含5A的50mL圆底瓶中加入3-叔丁基水杨醛(0.11g,0.6mmol,2.5equiv.)、无水MgSO4(0.17g,1.44mmol,6.0equiv)、CH2Cl2(20mL)、Et3N(0.15g,1.44mmol,2.0mL,6.0equiv.),室温搅拌反应过夜,TLC监测反应。反应完毕,将亮黄色固液混合物用短硅胶柱过滤以除去MgSO4和三乙胺盐酸盐,硅胶柱用石油醚/乙酸乙酯(2:1)洗脱至淋洗液无色,洗脱液减压浓缩得黄色油状物,用正己烷多次洗涤黄色油状物以除去残留的三乙胺盐酸盐,滤液合并,减压浓缩得亮黄色油状液体6A。
在含有化合物6A(0.18g,0.27mmol)的圆底烧瓶中加入甲醇(25mL),冰浴搅拌下先后加入NaBH4(0.10g,2.7mmol)和一滴盐酸,伴随气体产生的同时溶液由黄色变为无色,继续冰浴搅拌30min。缓慢加入稀盐酸溶液(2.0M)直至反应体系pH<1,以除去反应体系中多余的还原剂。反应体系用CH2Cl2(20mL×3)萃取,合并有机相,无水Na2SO4干燥,过滤,减压浓缩。所得固体用CH2Cl2/正己烷(1:8)重结晶得化合物7A。
推荐阅读:药理学医生怎么发表论文
3.结果与讨论
3.1.二胺的甲基化反应
伯胺的甲基化反应可用甲醛、硫酸二甲酯、碳酸二甲酯、甲醇等为甲基化试剂实现,但上述试剂参与的反应较难控制在单甲基化反应阶段。相比直接甲基化法,酰胺还原法在伯胺的单甲基化中有较好的应用。本论文采用Boc保护氨基,通过控制Boc2O用量、加料顺序和速度等可很好的抑制单边伯胺的双Boc等副反应,得到较好产率的化合物2A。再用LiAlH4氢化酰胺,可方便的在伯胺上引入一个甲基,得到化合物3A。化合物2A的单晶(图1)可通过乙酸乙酯重结晶获得。化合物3A的纯化可通过减压蒸馏(b.p.70℃,44Pa)实现。
3.2.氨基酚化合物的合成
由1,6-己二胺出发,经氨基与Boc2O作用的酰胺化、LiAlH4对酰胺的还原、仲胺与缬氨酸衍生物的酰胺化、酸性条件脱除Boc、游离氨基与叔丁基水杨醛的缩合及还原等反应可成功获得氨基酚化合物7A。但当以1,4-对苯二胺为原料时,在相同用料比的条件下,除得到了化合物4B和4B-1外,还分离纯化得到化合物4B-2。经对反应机理[13][14]和相关文献[15]的分析发现(路线2),化合物4B-2是L-缬氨酸在(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)活化作用下与1-羟基苯并三氮唑(HOBT)的缩合产物。化合物4B-1和4B-2的形成可能是由于化合物4B-1结构中芳香胺的亲核性较弱,使其较难进攻中间体化合物4B-2形成化合物4B。经研究发现,通过往反应体系中添加一定量的碱可促进化合物4B-1向化合物4B的转化。最后,通过调整L-缬氨酸的用量和添加胺用量可获得较高产率的化合物4B和4B-1。将化合物4B和4B-1进行后续反应,可分别得到化合物7B和7B-1。化合物7B-1的单晶(图2)可通过在石油醚/乙酸乙酯的混合溶剂重结晶获得。
3.3.化合物2A和7B-1的晶体描述
用R-AXISSPIDER的衍射仪上对化合物2A和7B-1的单晶进行测试,用石墨单色化的MoKα(λ=0.71073Å)辐射,对化合物2A在153(2)K温度下在2.64˚≤θ≤24.99˚范围内以ω-2θ扫描方式收集衍射数据。共收集到5447个衍射点,1562个独立衍射点(R(int)=0.0499)。其中1420个可观测点[I>2σ(I)]用于晶体结构解析,最终差值残余电子密度的最高峰为0.289e∙A−3,最低峰为−0.287e∙A−3。对化合物7B-1,在153(2)K温度下在2.65˚≤θ≤27.5˚范围内以ω-2θ扫描方式收集衍射数据。共收集到12,770个衍射点,5123个独立衍射点(R(int)=0.0357)。其中4778个可观测点[I>2σ(I)]用于晶体结构解析,最终差值残余电子密度的最高峰为0.14e∙A−3,最低峰为−0.16e∙A−3。化合物2A和7B-1的全部强度数据均经Lp因子校正,并做经验吸收校正。晶体结构由直接法解出,全部非氢原子的坐标及各向异性参数经最小二乘修正,用SHELXL-97程序(产生CIF的程序名称)对F2进行精修获得非氢原子坐标及各向异性参数,氢原子由差值Fourier合成和理论计算得到,他们的坐标和各向同性温度因子参与结构计算,但不参与修正。其晶体学数据列于表1。
4.中间体及产物表征
4.1.化合物2A
白色固体,收率89.2%;IR(KBr)/cm-1:3371(NH),1686(C=O);1HNMR(600MHz,CDCl3,ppm),δ:4.55(brs,2H,NH),3.03(m,4H,N-CH2),1.44(m,4H,CH2),1.42(s,18H,CH3),1.31(m,4H,CH2);13CNMR(150MHz,CDCl3,ppm),δ:156.1(C=O),79.1(CtBu),40.5(N-CH2),30.1(CH2),28.5(CH3tBu),26.4(CH2).HRMS(ESI)Calcd.forC16H32N2O4(M+H+):317.2396,Found:317.2537.131
4.2.化合物3A
无色液体,收率65.8%;1HNMR(600MHz,CDCl3,ppm),δ:2.45(m,4H,N-CH2),2.32(m,6H,N-CH3),1.38(m,4H,CH2),1.24(m,4H,CH2),0.89(brs,2H,NH);13CNMR(150MHz,CDCl3,ppm),δ:52.1(N-CH2),36.6(N-CH3),29.9(CH2),27.3(CH2).HRMS(ESI)Calcd.forC8H20N2(M+H+):145.1660,Found:145.1717.
4.3.化合物6A
亮黄色油状液体,收率58.6%;1HNMR(600MHz,CDCl3,ppm),δ:13.55(brs,2H,OH),7.99(s,2H,N-CH),7.31(d,2H,J=7.6Hz,Ar-H),7.20(s,4H,Ar-H),6.98(d,2H,J=6.0Hz,Ar-H),6.78(t,2H,J=7.6Hz,Ar-H),3.67(d,2H,J=7.6Hz,CH=N),3.31(S,6H,N-CH3),2.43(m,2H,CHiPr),1.41(s,18H,CH3),0.96(d,6H,J=4.4Hz,CH3iPr),0.82(d,6H,J=4.0Hz,CH3iPr);HRMS(ESI)Calcd.forC40H62N4O4(M+H+):663.4805,Found:663.4641.
澹版槑:鈶犳枃鐚潵鑷煡缃戙€佺淮鏅€佷竾鏂圭瓑妫€绱㈡暟鎹簱锛岃鏄庢湰鏂囩尞宸茬粡鍙戣〃瑙佸垔锛屾伃鍠滀綔鑰�.鈶″鏋滄偍鏄綔鑰呬笖涓嶆兂鏈钩鍙板睍绀烘枃鐚俊鎭�,鍙仈绯�瀛︽湳椤鹃棶浜堜互鍒犻櫎.

SCISSCIAHCI

