发布时间:2022-01-14所属分类:电工职称论文浏览:1次
摘 要: 摘 要:自放电是评价超级电容器性能的重要指标之一,显著影响超级电容器在实际使用过程中的能量转换效率。理解超级电容器的自放电机理,建立准确的自放电模型,从而开发针对性的改进方法,对提高超级电容器的实用性至关重要。然而,当前大量的研究工作集中于提高超级电
摘 要:自放电是评价超级电容器性能的重要指标之一,显著影响超级电容器在实际使用过程中的能量转换效率。理解超级电容器的自放电机理,建立准确的自放电模型,从而开发针对性的改进方法,对提高超级电容器的实用性至关重要。然而,当前大量的研究工作集中于提高超级电容器的能量密度和功率密度,对其自放电性能的关注较少。因此,本文综述了近年来超级电容器自放电研究工作的进展,着重介绍了电荷再分布、活化控制、扩散控制及电势驱动等自放电机制的数学模型及其影响因素,并从充电协议、表面化学改性、电极包覆、电解液/隔膜改性等方面总结了当前的自放电抑制策略,以期促进相关研究方向的发展。本文指出未来相关工作应在三个方面展开:首先,需要根据不同的应用需求建立完善的自放电评价体系;其次,需要将自放电的仿真模拟与先进的表征技术相结合,建立不同自放电机制的准确识别方法,并对其起因进行追溯;最后,需要根据自放电机制的不同,发展针对性的优化方法,实现自放电和其它电化学性能的同时优化。
关键词:超级电容器;自放电;多孔碳;双电层
电化学储能器件作为一种能量转换及存储的装置,是未来智能电网的关键组成部分,可有效解决风能、太阳能及潮汐能等清洁能源在时域和空间域上的限制,促进智能电子和电动汽车等新型终端应用的发展,因此,开发高性能的电化学储能器件是当前的研究热点[1-2]。电化学储能器件的关键性能指标包括能量密度、功率密度、循环寿命及能量效率等。其中,前三者最受关注,也是当前研究工作中着重提升的方向,这是因为它们与终端应用的用户体验直接相关[3-5]。以电动汽车为例,能量密度决定了电动汽车的续航里程,功率密度决定了电动汽车的制动能力及快充能力,而循环寿命决定了电动汽车电池组的维护周期及维护成本。与之相较,能量效率的关注度则较低,但实际上它对评价电化学储能器件的实用性至关重要。能量效率指电化学储能器件在荷电及开路状态下,贮存一定时间后,输出的能量与额定存储能量的比值,并主要取决于电化学储能器件的自放电行为[6-7]。严重的自放电会导致储存能量的迅速流失,降低能量转换效率,并影响储能模组的容量、配组和循环稳定性,缩短终端应用的待机时间。
在各类电化学储能器件中,超级电容器面临着最为严重的自放电问题[8]。超级电容器通过表面反应机制储能(电解液离子静电吸附于多孔碳表面,形成双电层(Electric double-layer)),不涉及电极材料的体相反应,可在数秒内完成充放电,具有高功率、长循环和高稳定性等优点,是工业节能、轨道交通和军工装备等领域重要的功率型储能器件[9-10]。然而,同样源于表面反应的储能机制,超级电容器的自放电现象严重[11],限制了其在上述领域中的应用效果。Smith [8]比较了商用超级电容器(2000F,Maxwell)和锂离子电池(2.4 Ah,E-One Moli Energy Corporation)在充电态下的自放电行为。结果表明,在 72 小时的开路贮存时间内,超级电容器的能量损失高达 22%,超过锂离子电池能量损失(3%)的 7 倍,其自放电问题可见一斑。因此,深入理解超级电容器的自放电现象,研究自放电的抑制方法,对提高超级电容器的实用性具有重要意义。
从本质上来看,超级电容器的自放电是其双电层中电荷自脱附的过程。超级电容器作为一个封闭系统,在充电态时具有远高于放电态的吉布斯自由能,因此在热力学驱动力的作用下,充电态的超级电容器趋向于朝放电态转变,即发生自放电。值得注意的是,理想极化电极所形成的双电层较为稳定,离子的自脱附需要克服较大的动力学势垒,因此通过对材料进行电化学电荷注入,形成双电层以改变材料的电子结构,可以作为调控材料光学、电学及磁学性能的有效手段[12]。然而,在实际的超级电容器中,由于电极材料、使用方式、微量杂质及电芯设计缺陷等因素的影响,电极往往处于非理想极化的状态,电极表面形成的双电层稳定性较差,电芯内部存在电荷通路,易发生严重的自放电现象。Conway [13]在上个世纪已对超级电容器的自放电现象进行了研究,并提出了数学模型用以描述其自放电过程。近年来,相关研究在自放电的机理解析、模型建立及抑制方法等方面已有了更为深入的认识[6],揭示了电场和浓度梯度力是自放电的主要热力学驱动力。这些工作对改善超级电容器的自放电性能,提高其实用性产生了良好的促进作用。本文对上述自放电的研究工作进行了回顾,以期促进该研究方向的进一步发展:首先介绍了不同的自放电机制、模型及影响因素,最后总结了自放电的抑制策略,并对未来自放电的研究方向进行了展望。
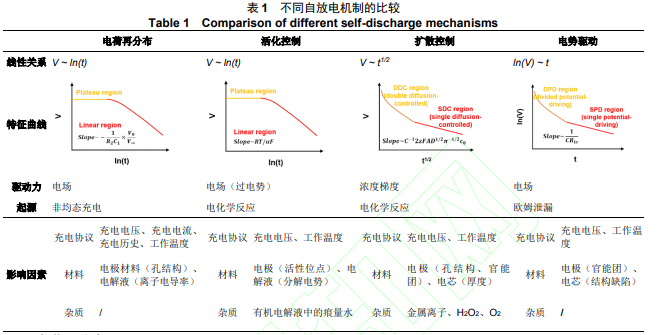
1 自放电机制及影响因素
双电层自放电的热力学驱动力来自于离子的浓度梯度力(∂𝑐⁄∂𝑥)和电势差产生的电场力(ΔV)。如表 1 所示,根据速控步骤的不同自放电机制可分为:电荷再分布(Charge redistribution)、活化控制模型( Activation-control model ) 、 扩 散 控 制 模 型 ( Diffusion-control model ) 和 电 势 驱 动 模 型(Potential-driving model);而根据反应类型的不同,自放电机制也可分为电荷再分布、欧姆泄漏(Ohmic leakage)和法拉第反应(Faradaic reaction)。深入理解不同自放电机制的起因、影响因素及行为特征,对针对性地改善超级电容器的自放电现象至关重要。
1.1 电荷再分布
电荷再分布发生在充电结束的初期,是电极和电解液界面处的载流子重新排列,使系统由非平衡态向平衡态转变的过程[14-15]。电荷再分布过程中系统没有电荷的损失,但电荷在重新分布的流动过程中,由于欧姆热效应,能量会以焦耳热的形式耗散,导致电压的迅速下降和储存能量的损失[16-18]。电荷再分布的形成主要源于非均态的充电,可通过动力学观点来解释。在充电过程中,由于电解液离子在电极中的有限扩散,电荷会优先流向具有快速响应特性的电极表面,而后流向于电极深处的孔隙,从而在电极内部产生了电势差,但测试电压反映的仅仅是电极表面的表观电势。因此,当充电结束时,电荷会重新排列使系统达到一个较低的平均电势。Kumar [15]采用基于第一性原理的一阶输运模型对上述过程进行了深入分析,发现电荷再分布实际上是在两个时间维度下进行。除了电势差驱动下的电荷再分布(数秒内),非均态充电过程中产生的离子浓度梯度也会作为驱动力,引发一个较为缓慢(数百秒)的电荷再分布过程。
影响超级电容器自放电的主要因素可以分为三类,即充电协议(充电方式、充电电流、截止电压、充电历史、工作温度)、体系材料(电极材料、电解液、隔膜)和体系杂质(水分、金属杂质、气体等)。对于电荷再分布,从其产生机制可以看出,与反应动力学相关的限制因素(充电协议和体系材料),将对其电压衰减过程产生重要影响。在充电协议方面,Kaus [18]详细研究了在不同使用情况下,商用超级电容器(Nesscap 600F)的电荷再分布过程。首先,降低充电电流密度,延长充电时间,可显著抑制电荷再分布现象的产生。在 15 分钟的快速充电条件下,电荷再分布导致的电压衰减可占据总漏电压的 50%(48 小时的开路测试);而将充电时间延长至两天可以消除电荷再分布的影响。这是由于低充电电流密度确保了电解液离子具有充足的时间迁移至电极材料的内部孔隙,从而使不同孔隙之间的电荷密度达到平衡。同理,在恒流充电后,对超级电容器施加一定时间的恒压充电处理也可抑制电荷再分布所致的电压衰减,并且已经在超级电容器的标准评价及实际使用中运用[19]。其次,提高超级电容器的截止电压,会导致更为剧烈的电荷再分布现象。该现象的物理意义在于,较高的充电电压可提供足够的电场驱动力,使电解液离子进入低电压下难以到达的内部孔隙,从而提高了电极材料的有效电荷存储面积,产生了新的 R/C 分支回路。这也反映在许多超级电容器工作的测试结果中,即电极材料的实际比电容具有一定的电压依赖性,充电电压越高比电容越高[20]。最后,一般来说工作温度升高会提高体系的电化学活性,从而加速自放电,但最近的研究表明温度对电荷再分布的影响实际上较为复杂。一方面,提高温度可以降低电解液的黏度,提高离子电导率,从而提高电极材料的反应动力学,降低时间常数,使电极更快达到平衡态;另一方面,它同样增强了界面离子的活性,降低了双电层的稳定性。因此,在不同的截止电压区间内,温度对电荷再分布的速率会产生相反的影响。
在体系材料方面,电解液离子在电极材料纳米孔隙中的传输速率是关键的动力学速控步骤,显著影响超级电容器的电荷再分布,并主要取决于电极材料的孔结构,包括孔径大小、孔径分布、孔径长度及孔型等[15,21]。对上述构效关系进行深入研究也有利于为电极材料孔结构的合理设计提供指导,以优化超级电容器的倍率性能[22]。一般来说,微孔(Micropores)具有远高于中孔(Mesopores)和大孔(macropores)的时间常数,意味着它需要更长的充电时间来达到平衡态[15]。然而,尽管微孔不利于离子的快速输运,但它却是比电容的主要贡献来源[23]。因此,需要对电极材料的孔径分布进行合理的设计,以在能量密度和功率密度之间达到平衡。为了进一步分析孔结构对电荷再分布的影响,Andreas [21]采用基于 de Levie 线性传输模型的孔隙模型,对不同孔型(锥形孔、倒锥形孔和圆柱孔)的电荷再分布现象进行了研究。在总电阻和电容相同的情况下,他发现孔长对各类孔型纳米孔的电荷再分布的影响是一致的,即孔隙越长电荷再分布所导致的电压衰减越大。此外,孔口处的孔径大小对电荷再分布起到关键作用,孔口尺寸较大的锥型孔对电荷的积累最为有利,这一点在平均孔径较小时尤为突出。这也意味着在孔口尺寸较小时提高材料的比表面积(增加孔隙长度),对提高超级电容器的能量密度是没有意义的,因为孔隙在充电过程的堵塞或离子的过量积累,会显著降低电极材料的有效电荷存储比表面积,并引发严重的电荷再分布现象。
1.2 活化控制模型
活化控制的自放电是法拉第反应型自放电的一种。在此过程中,双电层中发生法拉第反应,从而消耗其中的存储电荷,导致电压下降。与电荷再分布不同,上述过程中的储能系统会有存储电荷的损耗,造成贮存能量的流失。活化控制自放电的反应物质主要为电极界面处高浓度的杂质或电解液。当电极电势高于(低于)上述物质的氧化(还原)电势时,副反应会在过电势的驱动下发生,直至过电势随着反应的进行降低到 0 V[24]。最常见的活化控制自放电包括[25]:对超级电容器进行过充而引发的电解液分解,有机体系多孔碳中的吸附水在高工作电压下的分解,碳表面氧化官能团在水系电解液中的氧化。值得注意的是,上述法拉第反应过程通常伴随着电极材料的钝化和器件内压的增强,易引起超级电容器性能的下降和安全隐患。
1.3 扩散控制模型
扩散控制的自放电也属于法拉第反应型自放电,但与活化控制自放电不同之处在于,扩散控制自放电的驱动力为浓度梯度力,这是因为引发扩散控制自放电的物质为体系中的微量杂质。此时,浓差极化取代电化学极化成为该自放电副反应的关键速度控制步骤,即为了保证自放电的持续进行,电解液中的反应物质需要在浓度梯度力的驱动下,扩散至电极表面进行反应,从而消耗双电层中的储存电荷。该类自放电的常见反应物质为电解液中的金属离子、溶解氧(O2)及过氧化氢(H2O2)等[31-32],并通常作用于低于电解液分解电压的整个工作区间。值得注意的是,上述物质可能最初仅存在于电极材料中,而在储存和循环过程中才溶解进入电解液,因此除了电解液,在电极材料的备料过程中,也应减少上述杂质的引入。此外,扩散控制的自放电易引起连锁反应,产生“穿梭”的自放电现象[31]。
扩散控制自放电数学模型的建立为分析其自放电速率的影响因素提供了指导,可以看出电压衰减速率(曲线斜率)与扩散常数(m)相关,并取决于电容(C)、超级电容器厚度(h)、扩散系数(D)及初始反应物质浓度(c0)等。然而,上述数学模型没有反映出自放电速率与超级电容器的充电协议之间的关系。实际上,截止电压及工作温度与扩散常数(m)密切相关。首先,截止电压显著影响反应物质的初始浓度,这是因为浓度梯度的建立主要发生在超级电容器的充电过程,即杂质离子随着充电的进行不断在电极表面或近表面积累。研究表明,当充电电压超过一定阈值后,扩散常数与其呈现出显著的依赖关系,充电电压越高(电场驱动力越大),初始反应物质浓度越高,扩散常数越大[29]。其次,扩散控制自放电作为一个速率反应过程,其自放电电流与温度之间同样近似地遵循阿伦尼乌斯公式,即 log(i)与 1/T 呈线性关系,温度越高自放电电流越大,但与活化控制自放电不同,扩散控制自放电的活化能仅为 16~20 kJ/mol[13],代表杂质离子在电解液中的扩散势垒。造成该现象的原因可能在于提高温度可以增加电容量,降低扩散阻抗,增加反应物质的初始浓度,从而提高扩散常数,但上述三者中的关键项尚存争议。Ricketts [29]认为电容量与温度的依赖性较差,D1/2在高温时的增量不足以解释自放电速率的显著增加,因此温度对初始浓度的作用可能是其影响自放电速率的关键项。
在体系材料方面,首先隔膜的厚度对扩散控制自放电具有显著影响。增加隔膜厚度(超级电容器厚度)可以提高反应物质的扩散距离,从而降低自放电的浓度梯度驱动力。其次,电极材料的孔结构也是重要的影响因素[32]。对于微孔材料(活性碳),电极材料的比表面积及其比电容对自放电速率的影响较小。这是因为在高面积/孔容比的情况下,微孔中的反应物质会在最初的自放电阶段耗尽,且不会作为后续扩散自放电的反应场所。因此,公式中的 A 约等于电极的表观面积。而对于中大孔材料,扩散控制自放电根据孔径的不同,可能在时间维度上裂解为多个过程。Lu [33]研究了孔径为 20~50 nm 的多壁碳纳米管的扩散控制自放电现象,发现由于束间孔和束内孔对离子的传输阻力不同即 D 不同,扩散自放电可发生在束间孔和束内孔的表面,且两者具有不同的时间常数和衰减速率。
1.4 电势驱动模型
电势驱动的自放电属于欧姆泄漏,驱动力来自于正、负电极之间的电势差。从等效电路的角度看,在电势驱动的自放电过程中,电容单元并联了一个泄漏电阻;充电完成后,储存电荷通过泄漏电阻自发地释放,造成电压的衰减和能量的损耗[13]。泄漏电阻的产生源自超级电容器的结构缺陷,即正、负极之间存在内部的微短路通道,例如不适当的固定正、负电极导致两者直接接触,或流体电容器中正、负电极浆料间的分离膜发生破损。因此,可以通过评估泄漏电流的大小来检查超级电容器的结构可靠性。
电势驱动自放电的影响因素包括外部因素和内部因素。其中,外部因素主要为超级电容器的结构设计参数及工艺流程等,不属于本文的讨论范围。而内部因素(使用情况、体系材料)的研究对于深入理解该自放电过程及进一步的优化至关重要。从本质上来说,与法拉第反应型自放电类似,电势驱动自放电仍然属于一个速率反应过程,近似地遵循阿伦尼乌斯公式,因此该过程同样具有一个反应活化能。
2 自放电的抑制方法
对各类自放电现象的影响因素进行针对性的调控,可有效抑制对应的自放电现象。然而,在实际的超级电容器中,由于多种自放电机制的共同存在,对单一影响因素的调控可能同时作用于多种自放电机制,并产生互为矛盾的结果。例如,Beguin [36]发现尽管提高活性碳的中孔含量可以缓解电荷再分布现象,但其增加了电解液氧化还原物种与电极的接触面积,加剧了扩散控制的自放电现象,最终导致更大的电压衰减。此外,在抑制自放电的同时,也应以尽量保持其它关键性能指标(能量密度、功率密度、循环性能)为前提,并综合考虑成本的影响。目前,工业方面主要从工艺过程出发,减少电芯制备过程中的杂质引入,同时对电芯结构进行合理的设计,降低内部微短路的风险。而科研方面则从自放电的本质机理出发,发展协同的抑制策略。这些研究工作解析了体系与自放电的构效关系,有效的抑制了自放电,为工业生产提供了新的解决方向。本节对目前已报道的自放电抑制策略进行了总结。
2.1 充电协议
从前述讨论中可以看出,充电协议对各类自放电机制均会产生影响,因此优化超级电容器的充电协议以降低自放电过程的电压衰减是十分具有吸引力的,因为其避免了引入额外的材料或电芯生产的流程。然而,在实际应用中,该方法主要的抑制目标仅为电荷再分布产生的电压衰减,这是因为电荷再分布对充电协议的依赖性较大(截止电压、电流密度、充电方式、充电历史等),而活化控制、扩散控制及电势驱动仅受截止电压的影响。对于后者,毫无疑问我们难以通过牺牲截止电压的方法,来缓解自放电现象。这是因为截止电压通常需要设置在低于电解液分解电压(水系 < 1.23 V,有机系 < 2.7 V,离子液体 < 4 V)的最大值[9],以在避免电解液分解导致循环性能快速衰退的情况下,达到最大的储能密度(E=1/2CV2)。值得注意的是,该稳定电压也是活化控制自放电的“开关”,即在稳定电位窗口内通常不会发生活化控制的自放电现象。因此,学者提出可以通过对自放电机制进行解析,从而确认超级电容器的稳定电位窗口[36]。而对于电荷再分布,如前所述,抑制其电压衰减的关键是需要对超级电容器进行完全充电。在实验室中,该目标可以通过低电流的恒流充电、恒压充电及恒流脉冲充电等充电方式实现,但显然在实际应用过程中不会有长时间的充电过程,因为这会丧失超级电容器快速响应特性的优势。此外,过长的恒压充电也会导致能量效率的较大损失。目前,实际应用中的充电方式主要为恒流/恒压耦合充电,其设计关键在于如何分配两者的控制时间,以在充电效率和自放电之间取得平衡。在这方面,Wang [37]开发了一个超级电容器的物理模型,通过 Smoluchowski 漂移/扩散方程求解超级电容器电极内固相电荷的非线性分布情况,成功量化了充电电流和恒压保持时间对电荷再分布所致的自放电的影响,从而解析了恒流充电和恒压充电之间的耦合关系。该研究表明超级电容器模型的建立及其参数研究,是求解耦合充电协议最优解的有效方法;研究结果指出只有在总充电时间大于 1200 秒时,任何持续时间的恒压充电才会对电荷再分布的抑制产生有益作用;否则,对于更短的总充电时间,应使用尽可能长的恒流充电过程(即尽可能最低的充电电流)。
2.2 表面化学改性
碳材料表面的氧化官能团对自放电的加速作用已在多种电极和电解液体系中被观察到[38-40],从第二节的讨论中可知,其主要影响活化控制和电势驱动的自放电过程。此外,官能团的分解产物也可能引发扩散控制的自放电。因此,对多孔碳进行纯化处理,降低碳材料的表面氧含量是提高其自放电过程中的电压保持率的有效方法。例如,Wang [39]采用熔盐法(LiCl,KCl,KNO3)对还原氧化石墨烯进行处理(600 ℃),获得了相较于传统热处理的还原氧化石墨烯,氧含量更低的样品(2.3 at.%)。该样品所组装的超级电容器在离子液体(EMIMBF4)中展现出了更低的自放电速率,其电压由 3 V 衰减至 2 V 的所需时间为 0.9 小时,远高于传统热处理的还原氧化石墨烯基超级电容器(0.36 小时)。然而,氧化官能团对于超级电容器性能的影响不只是负面的,相反地,适量的氧化官能团已被证明可以增加电解液的润湿性,引入额外的赝电容量,提高孔隙的利用率[41]。因此,上述纯化处理难以避免会在增加材料制备成本的同时,降低其比电容和倍率性能。进一步的研究为解决上述矛盾提供了新的思路,Chen [40]通过氢还原处理制备了不同表面氧化官能团的淀粉基活性碳,并测试了其在有机电解液(SBPBF4 in PC)中的自放电性能。结果表明,不同的氧化官能团种类对自放电的作用不同,羧基(-COOH)和 RCOOR 会加速自放电而稳定性较高的-C=O 则对自放电影响较小,这说明可以通过官能团的精细调控获得兼具低自放电、高容量的电极材料。例如,研究表明,对于水系电解液中碳的表面氧化(> CxO + H3O + + e − => CxOH(H2O)),在最初的循环过程中,随着碳表面的逐渐氧化,不稳定的羧基会转变为羟基和羰基(OC=O→C-O/C=O),自放电速率大幅下降,因此可以通过对超级电容器进行简单的预循环处理以消除该自放电的影响[41-42]。此外,多项工作表明对碳材料进行氮掺杂,增强离子与电极表面的静电力,提高电极表面的电化学稳定性,也可有效抑制自放电[43-45]。
2.3 电极包覆
采用绝缘层对电极材料表面进行包覆已被证明是抑制自放电的有效策略,其可以有效阻碍电极/电解液副反应的发生,抑制法拉第反应型的自放电(活化控制和扩散控制)并同时提高超级电容器的工作电压[46-49]。然而,该类方法易引起超级电容器比电容及倍率性能的下降。这是由于绝缘层的形成易造成孔隙的堵塞,增加电解液离子的迁移阻抗。例如,Tevi [46]通过在活性碳上电化学沉积 PPO 阻塞层,成功将超级电容器的泄漏电流减小了 78%,但与此同时超级电容器的比电容下降了 56%。因此,如何调控绝缘层的性质以同时实现低自放电速率和高电化学性能是该类策略的关键。首先,研究表明,采用原位的电化学聚合/沉积策略是实现上述目标的必要途径,其有利于在多孔电极的表面形成均匀的包覆层以避免孔隙的堵塞。Jung [49]比较了在活性碳表面,通过自由基聚合及电化学聚合两种方式,形成 2-异氰基甲基丙烯酸酯包覆层的活性炭基超级电容器的电化学性能,发现电化学聚合的方法可以在抑制自放电的同时,保持较高的比电容量。另外,Li 认为该类包覆层的性质应接类似于固态电解质界面膜(SEI/CEI),即具有薄层(纳米级)、离子导通和电子绝缘的特性[47]。他通过在有机电解液中(1 M LiPF6 in EC/DMC)加入成膜添加剂(双氟草酸硼酸锂),利用其在 1.7 V vs. Li/Li+的电化学还原反应,在石墨烯表面形成了纳米级的固态电解质界面层,成功将电极的稳定电位窗口内由 1.5~4.3 V 拓宽至 1.1~4.3 V,并在保持比电容及倍率性能的同时,有效抑制了自放电现象。他也指出固态电解质界面层的形成除了可抑制扩散控制的自放电现象外,基于其去溶剂化效应,还可增强离子与电极间的静电作用力,形成更为稳定的双电层,从而抑制电势驱动的自放电现象。——论文作者:王宇作 1,2,卢颖莉 2,邓苗 3,杨斌 4,于学文 2,荆葛 2,阮殿波 5,6*
SCISSCIAHCI