0
发表咨询在线!

发布时间:2020-01-22所属分类:医学职称论文浏览:1295次
摘 要: 摘要:青钱柳是一种民间常用中药,青钱柳叶中有多种生物活性的次生代谢物,如有机酸、黄酮类、皂苷类、铱类、精油、无机元素等,但对于青钱柳次生代谢产物合成的分子机制仍未见有报道.使用Illumina公司Hiseq4000平台对青钱柳叶进行转录组测序,对reads进行拼

摘要:青钱柳是一种民间常用中药,青钱柳叶中有多种生物活性的次生代谢物,如有机酸、黄酮类、皂苷类、铱类、精油、无机元素等,但对于青钱柳次生代谢产物合成的分子机制仍未见有报道.使用Illumina公司Hiseq4000平台对青钱柳叶进行转录组测序,对reads进行拼接,得到50126个unigenes平均长度为1247bp,有19875(39.65%)unigenes由NCBI非冗余数据库进行注释的,23716(47.31%)unigenes被注释到GO数据库,14950个(29.82%)unigenes匹配到COG功能组.进一步的分析结果显示,6012(11.20%)个unigenes被富集到254个KEGG代谢通路,其中1212个unigenes参与了次生代谢产物的生物合成,并且发现126个unigenes参与异戊二烯代谢,包括萜烯类、香茅醛、呋喃酮、苷的代谢途径.此外,总共检测到21089个简单序列重复(SSRs).通过对老叶与嫩叶的转录组比较分析结果显示,在青钱柳不同生长发育时期的叶片中,基因表达上调占主异作用的过程主要涉及萜类和多肽的代谢、辅酶和维生素的代谢和氨基酸代谢,而基因表达下调占主异作用的过程主要涉及信号传输、类脂物代谢与能量代谢.该转录组数据可为青钱柳重要生物活性成分的生物合成和调控提供参考.
关键词:青钱柳;转录组;功能注释
青钱柳(Cyclocaryapaliurus)是我国特有的青钱柳属(Cyclocarya)植物,是一种主要生长在中国南部山区的著名中药.青钱柳叶常被用于治疗高血糖和高血脂[1-3],青钱柳叶具有抗抗氧化、调节血糖以及修复和保护胰岛等多种药理活性[4].大量的研究表胆,在青钱柳叶存在有一系列的有重要药理活性的次生代谢物,包括黄酮类化合物、总三萜和青钱柳甙Ⅰ、青钱柳酸B和阿江榄仁酸[5-6].近年来有各种各样的青钱柳茶被研发,青钱柳叶有效成分的开发利用成为了一个热点话题,而青钱柳是高大落叶乔木,青钱柳叶采摘难度大,青钱柳资源日逐匮乏.因此,对影响青钱柳叶次生代谢产物的活性成分的因素进行调控以提高活性成分含量,通过基因工程和细胞工程技术获得更多的青钱柳活性成分,这是当前缓解青钱柳资源短缺和需求逐年增加的矛盾的主要途径.然而,有关青钱柳次生代谢产物合成机理相关的研究极少,至今有少量有关运用ISSR和SSR标记对青钱柳种质资源进行遗传多样性分析的报道[7-8],由于基因组信息太少,很难在分子水平上对青钱柳次生代谢机制和调控进行更多的研究.
近年来,RNA-seq技术可用于预测基因或亚型的表达、检测差异表达的基因[9],现已被广泛应用于全基因组转录水平的量化,以及分子标记的挖掘和药用植物中各种次生代谢物生物合成相关基因的鉴定,如兰花、板蓝根、山茶等[10-12].对于至今仍没有基因组序列的物种,RNA-seq(高通量RNA测序技术)为人们进行次生代谢相关研究提供了一种可行的方法.
在本研究中,利用IlluminaHiseq4000平台获得了青钱柳叶的高质量转录组数据,共生成50,126个装配的unigenes,并对公共蛋白数据库进行注释,然后进行GO、COG、KEGG分类,检测到21089例假定的简单序列重复(SSRs).这些转录组数据提供了一个有价值的公共基因组资源,对于理解青钱柳叶次生代谢机制,促进发现与次级代谢途径及其调控有关的基因,以及未来青钱柳的基因表达谱和功能基因组研究都着十分重要的作用.
1材料和方法
1.1植物材料和RNA提取
2017年6月,在湖南通道县木脚青钱柳生产合作社基地采集7年生青钱柳老叶(枝条顶端倒数的第6~8片绿叶)与嫩叶(新冒芽的两叶一心),每个样品来自3个不同的单株.收集到的样本立即在液态氮冷冻、储存在-80℃.使用mirVana试剂盒(Ambion公司)提取总RNA.使用Agilent2100生物分析仪(AgilentTechnologies,SantaClara,CA,USA)评估RNA完整性,RNA完整性较好的样品(RIN≥7)用于进行后续分析.
1.2cDNA文库建设和Illumina测序
采用TruSeqStrandedmRNALTSamplePrepKit(Illumina)试剂盒准备转录组库.利用Oligo(dT)磁珠从总RNA中分离出poly(A)mR-NA,进一步纯化后,在加入缓冲液,将mRNA随机分为100~400b的短片段,以这些短片段作为模板,利用逆转录酶和随机引物合成第一链cDNA.随后,采用QubitadsDNAAssay(LifeTechnolo-gies)试剂盒,利用随机的六聚物引物(Illumina)合成了第二链.采用AgencourtAMPureXPkit(BECKMANCOULTER)的试剂盒对cDNA片段进行纯化和转换,进行末端修补,添加A尾混合物,充分混合,加连接混合液,30℃温浴10min,加反应停止缓冲液停止反应,增加PCR反应按如下程序进行:98℃30s;98℃10s,15个循环;60℃30s;72℃30s;72℃5min;10℃保存.加入AM-PureXP珠进行纯化.1μL示例加载在安捷伦2100生物分析仪检查库的大小和纯洁.使用Illu-minaHiseq4000平台从5’端到3’端对双末端RNA-seq测序库进行测序.
1.3数据筛选和重新组装
采用IlluminaHiSeqTM2500测得的数据称为原始读取,随后进行原始读取的质量控制,以确定测序数据是否适合后续分析.在质量控制后,通过筛选得到理想的读取,然后将25万对读取与NR在线数据库进行污染检测,以确定样品是否被污染.随后对合格的样品进行基因表达与基因注释分析,包括借助NR库、SWISSPROT库、KOG(真核)/COG(原核)等进行GO分析、KEGG代谢通路分析、基因表达结果筛选.以不同表达的基因为基础,对样品间差异表达的基因进行了GO功能显著富集分析与通路显著性富集分析.采用NGSQC4Toolkit[13]软件进行质量控制,去掉接头,过滤出低质量的碱基,最终得到高质量的干净读取.使用Trinityrnaseq_r20131110)[14]软件通过配对剪接法获得转录序列,然后使用TGICL[15]软件进行聚类、剔除冗余和扩展,得到最后一组unigenes,这是后续分析的参考序列,采用BLAST算法进行遗传相似性比对主要[16].
相关知识推荐:发表植物转录组测序类文章的SCI期刊
SCI是国际期刊,对投稿论文要求极高,再加上语言差异,一般在SCI上发表论文的作者很少,所以研究植物的作者,对SCI期刊了解得不是很多。下面小编就给大家推荐几本有关植物转录组测序类SCI期刊。
转录组数可以进行蛋白质功能注释,SWIS-SPROT注释,KOG功能注释,GO分类,KEGG代谢通路分析.unigenes序列采用blastx方法与NR、SWISSPROT、KOG库进行比对,得到与给定的unigenes序列相似性最高的蛋白(域值E<1e-5).unigenes的KEGG标注信息是使用KAAS(http://www.genome.jp/kaas-bin/kaas_main)获取的,基于SWISSPROT标注结果与GO信息,最终得到蛋白功能标注信息.
1.4unigenes的表达丰度计算和差异分析
在转录组测序分析中,通过对unigenes的计数来估计基因表达水平.unigenes计数与基因的真实表达水平呈正相关,但也与基因的长度和测序深度呈正相关.通过拼接得到unigenes文库,通过序列相似性比较的方法得到每个样本中每个unigene的表达丰度.借助在线软件bowtie2[17](http://bowtie-bio.sourceforge.net/bowtie2/manual.sht-ml)和eXpress[18](http://www.rna-seqblog.com/express-a-tool-for-quantification-of-rna-seq-data)进行差异比较.采用FPKM方法[19]计算uni-genes的表达数.根据DESeq[20]软件(http://bio-conductor.org/packages/releasebiochtml/DESeq.ht)计算基因的差异表达量.采用二项负分布检验(NB)检验读取次数的差异显著性.
1.5SSR标记检测
SSR是指基因组中的简单序列重复,SSR是在DNA在复制或修复的过程中,由于DNA的滑移而发生交换、错配,或者是在有丝分裂或减数分裂的过程中由于姐妹染色单体异质性交换的过程中而产生的简单序列重复.SSR标记在不同物种、同一物种不同位点,甚至是同一位点的不同等位基因之间都有可能存在比较大的差异.然而,即使SSR分布于同一基因组的不同位置,但SSR两端的序列确是相对保守的单拷贝序列,可以对根据SSR两端的序列设计特殊引物对SSR进行PCR扩增,再经过电泳检测可以得到长度条带,最终可以通过SSR长度的差异比较不同种质的遗传多样性.采用MISA和Primmer3.0(ht-tp://pgrc.ipk-gatersleben.de/misa/)软件SSR标记检测.
2结果
2.1转录组测序产量与组装质量分析
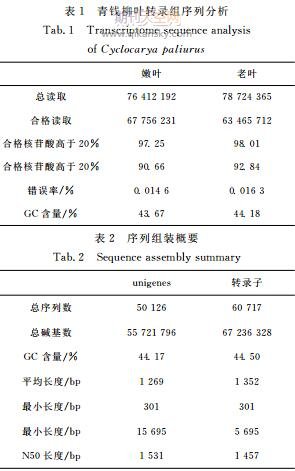
成功地获得青钱柳老叶与嫩叶的高质量RNA并分别构建了cDNA文库,分别采用IlluminaHiseq4000平台测序,分别得到76412192和78724365个读取(表1),从合格的读取中分别筛选到67756231和63465712合格读取,平均GC含量分别是43.67%和44.18%(表1).采用Trini-ty程序将所有高质量的读取组装成50126个uni-genes,覆盖50%所有核苷酸的最大unigene长度(N50)为247bp,unigenes平均长度为1169bp,构建了N50(1457bp)与平均长度1531bp的60717份转录子(表2),unigenes的平均GC含量为43.93%.此外,这些unigenes的长度在301~8456bp之间(图1),大部分在301~3800bp之间.50126个unigenes中,23563个(47.01%)小于400bp,6017个(12.00%)大于2000bp.这些unigenes可以进行注释.
2.2叶片转录组Unigene的功能注释
2.2.1叶片转录组Unigene的NR功能分类总共有19875(39.65%)unigenes注释在NRdata-base(表3).与此同时5572(11.11%)、17655(35.22%)、12465(24.87%)、15761(31.44%)、22135(44.16%)与3976(7.93%)个unigenes分别可以注释在KEGG、Swissprot、KOG、GO、Pfam与String数据库(表3).进一步的比对表明,注释的unigenes中92.14%的在NR数据中表现出为高度同源性(E值<1e-30),其中7.12%的E值为0(图2).此外,将带注释的unigenes与其他植物物种已知的核苷酸序列进行了比对,这些序列中12.68%能与葡萄已知的核苷酸序列最为匹配,随后是胡萝卜(8.32%),咖啡(5.37%),芝麻(5.20%)(图2).
2.2.2叶片转录组Unigene的GO功能分类利用Blast2GO[21]检索了青钱柳的GO术语,是将unigenes分门别类放入一个个功能类群.共26098个(52.06%)装配的unigenes被注释并分为3大类:生物过程、细胞成分和分子功能,然后分成64子类(图3).按生物过程分类,其中有细胞过程12139(45.25%),代谢过程22775(45.44%)和单个有机体过程21376(42.64%)是最明显的(图3),这表明这些unigenes在青钱柳代谢过程中扮演了一个重要的角色.在细胞成分分类中,unigenes主要与“细胞”21837(43.56%)和“细胞部分”14764(29.45%)有关,其次是“细胞器”8976(17.91%)和“膜”7699(15.36%).细胞外基质、线粒体相关复合体、突触、突触部分、病毒和病毒部分只分配了少量的unigenes.在分子功能类别中,大多数被显著地分为“结合”10876(21.70%)和“催化活性”9674(19.30%).在50126个unigenes中,11463(22.86%)被注释并划分为25个功能类别(图4).其次是“翻译后修饰、蛋白质转换、分子伴侣”2876(5.74%)、“信号转导机制”2354(4.70%)、“翻译、核糖体结构和生物起源”1874(3.74%)、“碳水化合物运输和代谢”1572(3.14%),而仅少量的unigenes被归类为“细胞运动”23(0.05%)以及“细胞外结构”15(0.03%),此外,879(1.75%)个uni-genes被归类为未知功能.
2.2.3叶片转录组Unigene的KEGG功能分类从图5结果可知8365个(16.69%)的uni-genes主要是用高度相似酶的数目进行注释,并根据代谢途径可分为4个分支(图5),包括代谢、遗传信息加工、环境信息处理与细胞过程,并进一步分组为254KEGG通路.值得注意的是在图中显示24537个unigenes(48.95%)参与了代谢的,其中12182(24.30%)参与了次生代谢产物的生物合成,如碳水化合物代谢3321(6.63%)、氨基酸代谢1697(3.39%)和脂质代谢1472(2.94%).此外,图6显示了青钱柳最大的20个带注释的通路组.最具代表性的KEGG通路是“碳水化合物代谢”,其次是“信号转导”、“其他次生代谢产物的生物合成”、“脂质代谢”和“能量代谢”.如图6所示,有58个(0.13%)的unigenes被归类到“信号转导”通路.利用KEGG通路数据库对上述信息进行归类,对今后青钱柳的基因功能及其调控机制的研究具有重要意义.
声明:①文献来自知网、维普、万方等检索数据库,说明本文献已经发表见刊,恭喜作者.②如果您是作者且不想本平台展示文献信息,可联系学术顾问予以删除.

SCISSCIAHCI

