����ʱ�䣺2022-04-09�������ࣺũҵ���������1��
ժ Ҫ�� ժҪ ���Ų������IJ��Ϸ�չ��Խ��Խ�����ֵ�ȫ���������ݱ��ⶨ�㷺Ӧ�á��ڶ������������ݱ���ʽ������ͬʱ�����˺˻��������ݣ����������������Ҳ�dz���Ҫ����ͨ�������ȫ�����������г��˺˻���������Ҳ������������������У���δӺ�����ȫ��������������ȡ
����ժҪ ���Ų������IJ��Ϸ�չ��Խ��Խ�����ֵ�ȫ���������ݱ��ⶨ�㷺Ӧ�á��ڶ������������ݱ���ʽ������ͬʱ�����˺˻��������ݣ����������������Ҳ�dz���Ҫ����ͨ�������ȫ�����������г��˺˻���������Ҳ������������������У���δӺ�����ȫ��������������ȡ��ƴװ��������������в�����Ӧ�ó�Ϊ������������ڷ�������ѧ���Ŵ�ѧ��ҽѧ�ȷ�����о�����֮һ�����ڴˣ���ȫ��������������ȡ��������������еIJ��Լ���ص��������Ϸ�չ�����ݴ�ȫ������������ê�������� reads �ķ�ʽ�ͺ���ƴװ���ԵIJ�ͬ�����Է�Ϊ�вο�����ƴװ�����ʹ�ͷƴװ��������ͬƴװ���Լ�����Ҳ���ֳ����Ե����ƺ;����ԡ������ܽᲢ�Ƚ��˵�ǰ��ȫ�����������л����������������ݵIJ��Ժ�����Ӧ�ã�����ʹ������ʹ�ò�ͬ���Ժ��������������轨�飬����Ϊ�������������������ѧ������о����ṩ�����ϵIJο���
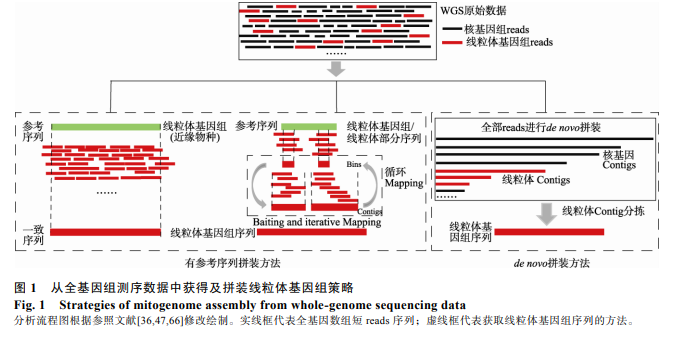
�����ؼ��� ȫ������;�����������;�вο�����ƴװ����;��ͷƴװ����;ƴװ����
���������������(mitochondrial genome)��Ϊһ������������ȡ���Ŵ���ǣ�����и�ͻ�����ʡ��������顢�߿�������ĸϵ�Ŵ����ص�[1]�����㷺Ӧ����ϵͳ��������������о�[2~5]��Ⱥ���Ŵ�[6~13]��ҽѧ[14~17]����̬ѧ�о�[18~20]�����������ڵ��о��Σ���������������еĻ�ȡ������ͨ��������ʽ��Ӧ(long range PCR, LR-PCR)�Ϳ�¡ PCR ������Ȼ����ͨ�����ﲽ��(primer walking)ɣ��(Sanger) �������ַ���ȷ�Ըߣ���ͨ���͡���ʱ�����ͻ��Ѹߡ����Ų������ą�չ���ر�����һ��������(next-generation sequencing, NGS)�ą�չ������ɱ��Ŀ����½���ʹ����������������еĻ�ȡ��ø�Ϊ���ס�Ŀǰ��NGS ������������(�� LRPCR �� NGS��RNA �����ȱ���(gap filling)��ֱ����ǹ������[21~23]��)ʹ�ø�ͨ�������Ϊ�ձ�������ȴ�ͳ�� Sanger ��������NGS ����ͨ���ߡ����Ը��������ø��͵Ļ��ѻ��ȫ����������(wholegenome sequencing, WGS)�����������кͻ���ת¼��[24]����һ���������Ļ���ԭ���ǣ�����ƽ̨�������� DNA ����봿����������� DNA �����ϳ� 50~700 bp �ĵ��� DNA �Ŀ�(DNA ����ȡ�����Ŀ��ƽ̨)���Խ���Ƭ�ε�����������ͷ��������������Ȼ��Բ����ļ��������� DNA ����މ�в���Ч��ȷ�����ٵػ�ô��� DNA ���У����ͨ��������Ϣ�����Ӻ�����ȫ�����������л�ȡ����������顣���������� Pacific Biosciences (PacBio) �� Oxford Nanopore �����Ӳ�����Ϊ�����ĵ��������������م�չ��������������މ�� DNA �������� PCR ���������Ҷ������ӵ���ʮ kb�������� 100 kb��ƴװ��õ�����������ȫ���������С������鼼���ą�չҲ��ʹ�������������ݱ���ʽ�����ӡ���ˣ�Խ��Խ����о��߳��Բ��ö����ͬ�IJ��Դ� WGS �����л�ȡ�����������[23,25~39]��
������ NGS ʱ����θ�Ч������������� DNA ������� DNA ����Ⱦ�����������������������Ĺؼ���Ŀǰ��Ҫ�������ַ�����ԣ�(1)�� NGS ����ǰ������ DNA ���������봿�������� DNA�����ֲ�����ͨ���Ȼ���ܶ��ݶ�����/�������Ļ����Լ��и������齫�� DNA�������� DNA����[40,41]��Ȼ���봿����������� DNA މ���Ŀ����ͨ������������ͨ���� NGS ����ǰ�ͽ��� DNA �������� DNA (��Ҷ���� DNA)���룬�Ա�֤��õ�������������������(��Ҷ����)���÷������������ڱ����˺� DNA ����Ⱦ��������������ת�Ƶ��˻��������(nuclear mitochondrial pseudogenes, Numts[42])�����ǣ��������봿���ķ������õ��Լ��мۖ����ȽϷ����ͺ�ʱ����������Ʒ������������Ҳ����һ����Ҫ�����Ŀǰ��Ȼ����������ս[43,44]���ر�������ϡҰ����������� DNA (ancient DNA, aDNA)���о��������Ϊ���ѡ�(2)��މ�� PCR����������������މ�� NGS ���ò������������������������������Ŀ��Ƭ�Σ��ٽ���������ֱ���ϻ�މ�� NGS �������蹹�� DNA �Ŀ�[45]���÷���������������Ҫ����ʼ DNA �������٣��ر��ʺ�С������ͻ��� DNA �о����ؼ�����ģ�� DNA �������� PCR ����������ԡ�
����NGS ���ݱ��㷺Ӧ����������ѧ�ĺܶ�������������މ������ѧ��Ⱥ���Ŵ�ѧ�Ƚ�ʾ���ֵ���Դ����ɢ��ʷ���慧������Ҫ�����á��о����dz������ֺ˻������ݺ����������ݱ��ֳ���һ�µ���ϵ��ϵ���ر��Ǿ��и��ӵ�Ⱥ����ʷ����Ⱥ(������������Ŵ�Ư�䡢ƫ����Ǩ���������ϵ�ּ��)���ɼ����ڷ��� NGS ����ʱ�����˺˻����������⣬���������������Ҳ�dz���Ҫ��Ȼ����Ŀǰͨ�� NGS������õ�ȫ�����������м���������������������ݺͺ˻��������ݡ���ȫ�����������У���Ȼ��˻��� reads �IJ��������ȣ������� reads �IJ�������Ǻ˻���� 100~1000 ��(ϸ���д��ڼ�ʮ�����ٸ�����) [46]������������������ܵ� reads ����ֻռ�� WGS �� reads ����һ���֣����ҳ����ܵ��˻����Ҷ����(��ɫֲ��) reads ����Ⱦ����ˣ�ʹ�ø�Ч��������Ϣ���ߺͷ������ԴӺ�����ȫ�����������п���ȷ�ػ������������� reads ������ȷ��މ�к��������������ƴװ���Ե÷dz���Ҫ[36]�����Ľ��ܽᵱǰ���õĴ� WGS �����л�ȡ��������������е�ƴװ���Լ�����������Զ�ʹ������ʹ�ò�ͬ���Ժ��������������轨�顣
����1 �вο�����ƴװ���Լ�����Ӧ��
�����вο�����ƴװ������Ҫѡ���Ե���ֵ�������������Ƭ����Ϊ�ο����д��о���Ⱥ��ȫ�����������в��������� reads�����ݴ� WGS �����в��������� reads �Ƿ���Ҫ�������������������Ϊ�ο����У�Ŀǰ���õIJ��Կ��Է�Ϊ��(1)���������������������ƴװ����;(2)����������Ƭ�ε�ƴװ����[47,48](ͼ 1)�������ݷ��������ϣ�����ʹ��ȫ������ȶԹ���(�� BWA[49])���� reads ӳ�� (mapping)��������ο������ϣ��������е������Բ��������� reads��Ȼ����ʹ�ò�ͬ�������ӳ����ԶԲ��������� reads މ���������죬ֱ���ӳ�������������������鳤�ȡ�
����1.1 ���������������ƴװ���Լ�����Ӧ��
���������������������Ϊ�ο����л�ȡ���ֻ�Ⱥ�����������������еķ������㷺Ӧ����ϵͳ������Ⱥ���Ŵ�ѧ�о����� Ko ��[50]���ִ����è���������������Ϊ�ο����У���ȡ��һ�� 2.2 ����ǰ����è������������顣��ԭ���Ǹ���ͬԴ�ȶԵ��о��������� WGS ����ӳ�䵽��Ե���ֵ�������������ϣ��ٸ��������� reads ����ص���꣬�Ӷ�������е��ӳ�(ͼ 1)�����ַ���������ȡ�Ͳο�������һ�µ�����(consensus sequence)������ȷ�Ըߣ������ٶȽϿ��Ҳ��ļ�����Դ��
�������Ų������ą�չ�������ݷ�������������Ҳ�����ӣ��ر�������������������о�����������މ����ʷ�����������弲���ȷ�����о�[51,52]���ƶ�������������������ƴװ��ע����������ą�չ(�� 1)��MIA �ǽ����������������������ƴװ���������о��߶��ᰲ���ع������ͷ�ᵽ�� DNA މ�и�ͨ����������ִ��˵��������������Ϊ�ο����У�ʹ�ø�������ȡ���ᰲ���ع�����������������[53]������������������������ݵIJ����ۻ����о�����IJ����������ݷ��������������Ĺ����������Ҫ��һЩ����� windows ͼ���û�������������㷺ʹ�ã����� MitoBamAnno-tator[54]��MitoSeek[55]��mtDNA-profiler[56]��mit-o-matic[57]�� MToolBox[58]��Phy-Mer[59]��mtDNA-Server[60]�� MitoSuite[61]�ȡ���������֧�ֶ��������ļ���ʽ������ mtDNA-profiler �� mit-o-matic �⣬����������֧�ֶ�މ�Ƶ� Bam ��ʽ�ļ�����ˣ���Щ��������ֱ�Ӷ�ȡ��ͬ������������ݣ��ӿ��������������̡�ֵ��ע����ǣ������������û�ѡ��IJο������������в��죬�� MitoBamAnnotator��mtDNA-profiler �� mit-o-matic ���ṩ�� 1 �����������(rCRS)�� MitoSeek (rCRS, hg19)��mtDNA-Server (rCRS, RSRS) �� MToolBox (rCRS, RSRS)�ṩ�� 2 ���������ݣ��� MitoSuite �ṩ�� 5 ������ο�������(rCRS�� RSRS��hg19��GRCh37 �� 38)��ʹ�� Phy-Mer �������û������Զ���ο����������С����⣬ͨ�� MitoBamAnnotator��MitoSeek��MToolBox��mtDNA- Server�� mit-o-matic �� MitoSuite �������û�����������Ӧ����(������С��λ����Ƶ�ʣ�MAF)����������������ı���λ���������λ��(heteroplasmic sites, �������������������ͬһ��λ�ô������ּ��������ϵļ�����ͣ���Դ��������Դ��Ⱦ�������������������������reads ƥ�����ȣ�Ҳ��������Դ������������)��MitoBamAnnotator ��Ҫ������Ԥ��������������λ��DZ�ڵĹ��ܣ���ʹ�ù��ܱȽϵ�һ��MitoSeek �� MToolBox ��չ�˷������ܣ����������忽����Ŀ���ȶ��������ṹ������ȹ��ܡ�MitoSeek �����Խ��� Circos[62]�����Լ����ı� �� މ �� �� �� �� �� �� �� �� �� �� �� �� �� (structural variations, SVs)�͵����������(single nucleotide polymorphism, SNPs)��MToolBox �������ڿ��Ե��η���������壬���ҽ�������Ϣ��¼�� VCF �ļ��У������ױ�������ע�͡����û��������з���Ƚϣ� MitoSeek �� MToolBox ��һ����� Perl ������Ե� Linux ���㻷����������Ҫ���ض�������� Perl ģ��ͱȶ�����(BWA)�Լ�����������(GATK[63])�����ڷ�������Ϣ�о��������û���װ��ʹ������������Խ����ѡ�mtDNA-Server �� mit-o-matic �����������û�ͼ�η������ߣ��û�����Ҫ���ӵİ�װ���̣���ͨ��ע���������ϴ����ݎ�މ�з��������������ݷ�����Լ�ȱ�����������ļ���С�����ƣ��ر��Ǹ߲�����ȵĸ����ϴ����ݽϻ��������ڿ����� MitoSuite ������չ�˸���ʵ�ù��ܣ����ܸ�ǿ��������������������ƴװ�������⡢��������ע�ͺ���Ԥ�⡢������Ŀ�����������ǶȵĿ��ӻ��ȡ�MitoSuite ������������ڵ�����������Ҫ��װ�������ӵļ���ģ�飬��ͼ�λ�����ϵͳ���ܱ������е�һ�����ײ���������������ֱ�Ӵ� Bam �ļ����Զ�����һ�������к�މ��ϵͳ������Ⱥ���Ŵ�ѧ���о�[61]�����Զ��������������������о�����ѡ�� MitoSuite ���������ơ�
��������������ʹ���������������������ȫ�����������л�ȡ��������������У����Ƚ���ȫ������ȶ��������������õ� BWA �� Bowtie/Bowtie2[64]�������� reads �в�������������� reads�������ֱȶ������������ڿ��Զ� reads ����� reads �ദƥ��މ��ɸѡ���ˣ�ͨ���������ʿػ�ȡ�������������� reads�����ǣ������� Numts �������忽�������Ӷ�Ӱ�������������Եļ�⡣���⣬��Щ���������������Ҫѡ���Ե���ֵ������������ο����У����ѡ��މ����ϵ�Ͻ������ֵ��������������Ϊ�ο����У���ȫ������ȶԵĹ����п��ܻᅧ�� reads ������������з�����²�������ȶԲ��϶�����ȱʧ����(gap)���Ӷ�Ӱ�쵽���������������ƴװ��ȷ�Ժ�������[38]����ˣ�ѡ��������ֵ��������������Ϊ�ο������Ǹ÷���������Ӧ�õĹؼ�������Ҫ�о���������ȷ�����Ե���֣�������ȷ�������Ե���ֵ�û��������������������ݵ�����£�����������кܴ�ľ�����[36,39]��
����1.2 ����������Ƭ��ƴװ���Լ�����Ӧ��
��������������Ե���ֵ�������ȫ��������Ϊ�ο����е�ƴװ���Լ���ص����������������˵������������ƴװ��������ͱ���ע�͵ȡ�����Խ��Խ���������ֵ��о�����������������Ҳ���㷺Ӧ���ڷ�ģʽ���ֵ��о���[65]�������˵Ļ�������Ϊ�ο����е���������ȡ�ͷ����������ֵ���������������оͱ��ֳ��ܴ�ľ����ԣ����������Ҫ�������÷�Χ����������������ƴװ����������reads ֱ��ӳ�䵽�����������ο����е�ƴװ�������ƣ�������ѡ���Ŵ���ϵ�Ͻ���Ͻ����ֵ�����������飬���������岿�����У���މ���������ֵ���������������л�ȡ��ƴװ���÷������Ƚ���ȫ������ȶ����������˺�� WGS ����ӳ�䵽�ο������ϣ��߸��Ƕ��������������� reads ������п�(bins)����Щ������ bins ���߸��� bins �ص�������ӳ� Contigs �滻ԭ�ȵIJο����У�����Ϊ�´�ӳ��İ�����(baiting sequencing)�����η����� WGS ����ӳ�䵽�����ɵİ��������ӳ����У�����ӳ�������������������鳤��(ͼ 1)������ӳ����滻�����п��Ա���ο����к�ƴװ������ƫ���ԡ�ƴװ��������Ҫ���� Kmerֵ(ƴװ������ reads��ϳɳ���Ϊ K ��һ�ι̶�����������)��С�������� WGS ����ӳ�䵽��������މ�������ӳ��������Ҫ���Ĵ����ļ�����Դ��ԭʼ����Խ��Խ���ļ�����Դ�����ѡ���Ŵ���ϵԽ�������ֻ�ѡ��İ�����Խ�̣�ƴװʱ�������ӳ�����Ҫ�����ѭ������������ʱ��Ҳ��Խ����
����Hahn ��[66]������ MITObim ��������ֱ�Ӵ� WGS ������ƴװ��ģʽ���ֵ�����������飬�������Ƕ���� MIRA �� IMAGE ����ģ�顣��� MIA�� MITObim ��ȷ�Կ��Դﵽ 99.5%���ϣ����ظ����������Ч��� gap�������ٶȺ��ڴ�����Ҳռ�����ƣ���ΪĿǰ��㷺ʹ�õ������������ƴװ��������������֧��˫������(paired-end reads, PE reads)��֧�� Iontorrent��454 �� PacBio ����ƽ̨���ݣ����ҽ���ԭʼ���� reads ������Ҫ���� 20~40 ����������������������ԭʼ reads �������ȡ���� reads�������ͼ��� reads �������������������ܻ�Ӱ��ƴװ�����ȷ�Ժ������ԡ���Ȼ��MITObim Ҳ����������������ƴװ��һЩ��Ϊ���ӵ����⣬�� Numts�����ӵ��������ֲ���������ƴװ��[67]��ARC[47]������ƴװ���������� MITObim ���������߶�����ѡ����Ե��ϵ�Ͻ������ֵ��������������������岿�����оͿ��Եõ���������������������У���Ҫ�IJ������������ӳ���ʽ�� ARC ��ֱ�Ӷ� bins މ��ƴװ������е��ӳ����� MITObim ���Ƿ������� reads ����������ӳ������ӳ����С��������ȫ������ƴװ������ARC ���ǽ��� reads މ�д�ͷƴװ��������ͨ��ӳ��ķ�ʽ�� reads �ص��� bins މ��ƴװ���������ڲ����ڴ棬�����ٶȽϿ졣���⣬ARC �����ϲ��ܽ������ص� DNA �����͵������� reads ��Ӱ�죬�ر��� aDNA�����������ٶȱ� MITObim �ʹ�ͳ��ƴװ������[47]�� Li ��[68]ʹ�� ARC ������ 19 �������߳�(Caenorhabditis)����މ�������������ƴװ�������˲�ͬ����ƽ̨(Roche��454��Illumina �� Ion Torrent)�������������ƴװ��Ӱ�죬������� ARC ������ 454 ƽ̨������މ�з���ʱ����������ܵ�ԭ�������г��ȷ�Χ�������ݷ�����Ҫ�ϴ�ļ�����Դ������ ARC ƴװ�������Զ�Ҫ�� MITObim �á�Ȼ���� Dierckxsens ��[47]�� ARC �����Խ���Ҷ����(Gonioctena Intermedia)މ�������������ƴװ��������־��� ARC ȷ�Ը�(99.99%)�������ܽ�������ƴװ��һ�� Contig �ϣ������Խϲ�(���ǵ������������� 85.39%)��
����Dierckxsens ��[38]������ NOVOPlasty ������������ SSAKE[69]�� VCAKE[70]�㷨���������� reads ����ڹ�ϣ���У��Ա� reads �Ŀ��ٶ�ȡ����������ٶȽϿ졣NOVOPlasty ������Ҫ�ṩһ�������У�������һ���� read��һ�α���������У���������������������������С�ֵ��ע����ǣ�NOVOPlasty �� ARC ƴװ���Բ�ͬ���ǣ�NOVOPlasty �����ṩ�İ����д� WGS �����л�ȡ������������һ�� read��Ȼ���ٶԲ��� read މ��˫�����졣���߽� NOVOPlasty �뵱ǰ������ƴװ������Ƚϣ����� MITObim��MIRA��ARC��SOAPdenvo2 �� CLCbio��������֣����� ARC �⣬������������������ƴװ��һ�� Contig��ͨ���� NOVOPlasty ƴװ��������މ������������û�Ѕ���ȱʧλ��Ͳ�ȷ���ļ��λ�㣬����ȷ�Ժ������Ըߡ�NOVOPlasty �ļ����ٶ���졢�����鸲�Ƕ���ߣ�CLCbio ȷ��ͬ��Ҳ�ﵽ�� 100%�����ǻ�����ĸ��ǶȲ���(89.96%)�� MIRA �� ARC ��������ߵĻ����鸲�Ƕȣ�����ȷ����͡����Ӳ��ǶȺ� reads �ij��ȿ������ NOVOPlasty �������Ժ�ȷ�ԣ��ر��Ǹ��ظ��� AT �����ߵ�����NOVOPlasty ���в���Ҫ��������������ģ�飬�����û���˵��װ�Ͳ����Ƚϼ�[38]��
����Ŀǰ����Ҷ���������ƴװ����ͬ���ʺ�������������ƴװ������ IOGA[71]��GetOrganelle[72] �� ORG.Asm[73]�ȡ�IOGA �� GetOrganelle ������ MITObim �е�“Baiting and iterative ӳ��”�������̡� IOGA ����������Ҫ Bowtie2��SOAPdenovo2��SPAdes 3.0[37]���������������������� reads��ƴװ���̻���Ҫ����ƴװ���� Kmer ��С(��ΧΪ 37~97)�����ͨ��ƴװ��Ȼ����(assembly likelihood estimation, ALE)�Ӻ�ѡ�� Contigs ������ȷ�������������[74]�����ַ����ʺϽ���̶Ƚϴ����Ʒ��������������Ҷ���������ƴװ�����粩�����Ʒ�ȡ�������ƴװ�����Ƚϣ�IOGA ʹ�� ALE ������ɸѡƴװ�õ� Contigs�����ͨ�������Ȼֵ���ж����ŵ�ƴװ���С�GetOrganelle �� IOGA ���ݷ������̷dz����ơ� GetOrganelle Ƕ���˶����� Bowtie2��BLAST[75]�� SPAdes 3.0 ����ģ�飬˫�� reads �͵��� reads (singleend reads��SE reads)��������Ϊ GetOrganelle �������ļ���GetOrganelle ����ֱ���� SPAdes ƴװ�Ĺ�����މ�� reads ��������ʹ�����ˣ������������� reads ��Ϊ������������ IOGA �� MITObim ����Ҫ����������������ǰމ�е����� reads �Ĺ��ˡ� IOGA �� GetOrganelle ƴװ������Ƕ�� SPAdes �������ģ�飬��ƴװ��������Ҫ�������� Kmer ֵ�Ĵ�С��ѡ����ʵ� Kmer �����ܹ���֤������ Scaffolds �� Contigs �������Ժ�ȷ�ԣ������Լ��ټ���ʱ��������ڴ�[72]��
������������ŵ����Ӳ��� PacBio �� Nanopore ��Ƭ�β������ą�չ��һЩ�������ֵ�ȫ���������б������Ӧ�ã��ر��Ƕ�����ֺ��ظ������֣���ʾ�˳�Ƭ�β�����������[27,76~80]��ͬʱ���Ѿ���������һЩ������ƴװ PacBio �� Nanopore �� reads ������������ HGAP[81]��Falcon (https:// github.com/PacificBiosciences/falcon)��Canu[82]�� Sprai[83] �ȣ�������Щƽ̨����õ��ij� reads މ���������Ҷ���������ƴװ�ķ������㷨����ȱ����Ŀǰ�Ѿ���һЩ�о���ֱ��ʹ�� PacBio �� Nanopore ƽ̨މ�����������������މ��ƴװ[25~29]��Soorni ��[84] ���� Perl ������Կ����� Organelle-PBA ֱ�Ӷ� PacBio ƽ̨����ȫ�����鳤Ƭ��މ���������Ҷ����������ƴװ��Organelle-PBA ��װ��ʹ����Ҫ��װ���� Perl ģ��Ͷ������������� BlasR[85]�� Samtools[86]��Blast[87]��SSPACE-LongRead[88]��Sprai �� BEDTools[89]�ȡ���Ȼ PacBio �� Nanopore ����ƽ̨���Եõ������� reads��������Ȼ����һ���ļ�������ʣ������Ҫʹ�ü����������މ�м������������ Sprai���� PacBio �� Nanopore ����ƽ̨����Ҫ�ڽ���Ĺ�����މ�� DNA �����Ϻ��������Ҿ��ж������ص㣬���Կ��������ý������������һ���Բ�ͨ����Ч������ Numts ����Ⱦ����ͬʱ��Ϊ PacBio �� Nanopore ����ƽ̨����Ʒ DNA �����м����ϖ��Ҫ��Ҫ��֤ DNA �������ԣ����� OrganellePBA ��ʹ��Ҳ�о����ԡ�
����2 ��ͷ(de novo)ƴװ���Լ�����Ӧ��
����Ŀǰ��������Խ��Խ������ֵ�ȫ���������ݺ���������������ݱ���������Ҳ�о���������ֵĻ�������Ϣ��δ���ⶨ�����û�вο����������е����֣���ͷƴװ��һ�ֿ��ٺ�ȷ�ػ�ȡ�Ŵ���Ϣ�IJ��ԣ����ַ������㷺Ӧ���� DNA �� RNA ����ƴװ�������������Ĵ�ͷƴװ��˻������ƴװ�������ƣ����ȴӺ�����ȫ�������������ҵ��� reads ��һ�������У�Ȼ���ٸ��ݲ�ͬ���ȵĴ�Ƭ���Ŀ�މ�� Contigs ����������ӣ�����ӳ��� Scaffolds ˮƽ������������ reads ����Դ��ͬ�����Է�Ϊ��ȫ�����������д�ͷƴװ�������������Ժʹ�ת¼�������д�ͷƴװ�������������� (ͼ 1)��
����2.1 ��ȫ�����������д�ͷƴװ�������������Լ�����Ӧ��
������ͷƴװ����������鷽������Ҫ�ṩ�����������������������岿��������Ϊ�ο����С���ͷƴװ���Ƚ� WGS ��ȫ�� reads މ�д�ͷƴװ[47,48]�������˻������������� reads ���ֱ�ƴװΪ��Ƭ�����У�Ȼ��������������������г��Ⱥ߲������މ���ϖ��Contigs���˵õ���ѡ������Contigs������� WGS ����ӳ�䵽��ѡ������ Contigs �ϣ������ӳ� Contigs��ֱ���ӳ�����������������鳤��(ͼ 1)�����е������� Norgal[36]�� MitoZ[39]�ȡ�����һЩû�н�Ե�������������������֣����� DNA �������ص���Ʒ(���� aDNA ����)�����вο�����ƴװ�������кܴ�ľ����ԡ����ԣ��� aDNA ������ DNA ����މ�� NGS ������މ��������������ͷƴװ����һ���ЁY��Ч�IJ��ԡ����ǣ����ַ�������Ҫ������ȫ�������ת¼��ƴװ�������ͼ���ģ��(���� SOAPdenovo2[90]��SPAdes[37]�� Velvet[91]��BIGrat[92]��CLCbio (https://www.qiagenbioinformatics.com/products/clc-assembly-cell)��SOAPdenovo-Trans[93]�� Trinity[94]��)����������������މ��ƴװ��������Ҫ�������� Kmer ֵ�ķ�Χ�Դﵽ��ѵ�ƴװ���������ԺķѼ�����Դ�������ٶȽ�����
������ͳ�Ĵ�ͷƴװ���������� SOAPdenovo2�� Newbler��SPAdes��Velvet��CLCbio��ALLPATHS[95] �� Platanus[96]�ȣ���ȫ����������ƴװ�����У��������� Scaffolds �� Contigs ���������˵�����ͷƴװ������������������ͳ�Ĵ�ͷƴװ�������ڷ��������п��������� reads �ĸ߲�����ȣ������ǽ���ɾ����Ŀǰ�Ѿ������දֲ���������������ô�ͷ��ƴװ�����������������������������С� Lee ��[97]�Խ۹��ƵĽ۹�(Platycodon grandiflorus) �͵���(Codonopsis lanceolata)މ���˵��ǶȻ��������Զ������������މ��ƴװ����������ʹ�� Celera��SOAPdenovo, SPAdes �� CLCbio �� 4 ��ȫ������ƴװ������ȫ�� reads މ�д�ͷƴװ���õ��ɺ˻������������ɵ� Contigs �⣬��θ���������� Contigs �ͺ˻������ Contigs ƽ��������ȵIJ���ȷ����ѡ������ Contigs���ٽ� WGS ���ݱȶԵ���ѡ������ Contigs �ϣ����ѭ����� Contig ���ӳ������õ������������������[97]������������ƴװ���ԣ�Al-Nakeeb ��[36]������ Norgal ��������ʹ�� MEGAHIT[98]ƴװ������ NGS ����މ�д�ͷƴװ��Ȼ���ٽ� NGS ��������ӳ�䵽ƴװ�õ� Contig �ϣ�ͨ��������ͺ˻������ reads ���Ƕ����ж������� Contig(s)������ͨ����������ͬ���Ե������������ƴװ�����Ƚυ��֣�Norgal ������ȷ�Ժ� NOVOPlasty �������ƣ����Ǵ������ٶ������Ƚϣ� NOVOPlasty ���� Norgal �� MITObim Ҫ�죬ԭ���� Norgal ��Ҫ������ͬ Kmer ��С������������މ��ƴװ��Ȼ���ٱȶ� reads �ͼ���˻����� reads �IJ���������ж�ƴװ�Ŀɿ���[36]��
�������֪ʶ�Ƽ��������������������ʲô�о��ɹ�
���������û������ݷ���������Խ��Խ��Ҫ�����Ч�ʵ����ݷ������̡�����ȫ������õ��û����������Խ��Խ��Ϊ���е���Ҫ��Meng ��[39] ������ MitoZ ��������“һ��ʽ”�ض������������މ��ƴװ��ע�ͺͿ��ӻ��������������˶��ּ���ģ�飬����ԭʼ���ݵ�Ԥ��������ͷƴװ����ѡ���������еĸ�����������������ע�ͺͿ��ӻ��ȹ��ܡ�����������������������ܶԵ������� reads���������ȱʧ�� reads �ͽ����� PCR ����� reads މ�й��ˣ��Ա�֤�����������ݵĿɿ��ԡ�MitoZ ������ SOAPdenovo-Trans ���㷨���Ӻ˻������е� reads މ�������������Ĵ�ͷƴװ����ԭ���ǣ���������������� reads ��ƽ��������Ƚ��Ⱥ˻�����ĸߣ����ò�ͬ�� Kmer �������ﵽ��ѵ�ƴװЧ������������ṩ������ƴװ��ʽ�����ģʽ(quick model)�Ͷ� Kmer ģʽ���������ߵĽ��龡����ʹ�ö� Kmer ģʽ������ͬ Kmer �������Ա�֤���������������ƴװ�������Ժ�ȷ�ԡ���ƴװ�Ļ������������е��ܳ��ȷ���މ�бȽϣ�MitoZ ���вο����е�ƴװ���Ը��������ƣ��ر��Ƕ������ּ����ƶȺܵ͵Ļ����⣬���˸��������㷨�IJ��죬�ظ����С�AT ��������������(������λ��ռ�ܱ���λ�������)��Ҳ��Ӱ��������������ƴװ�����Ժ�ȷ�ԵĹؼ�����[39]��MitoZ ��������������ע��(Blast��Genewise��MiTFi �� Infernal)�Լ����ӻ�(Circos)���ܼ������������������ģ�飬��˼�ӵ���չ��ƴװ�����Ĺ��ܣ�Ҳ����ؼ������ݵķ������̡�
����2.2 ��ת¼�������д�ͷƴװ�������������Լ�����Ӧ��
������һ���������ą�չͬʱ�ƶ���ת¼��ˮƽ���о�����ת¼�������л�û�������������Ѿ��ܳ��죬���ܵ� RNA ת¼���а���������������������ת¼���������о��߿����˿��Ը�Ч�ش�ת¼�������и������������������е�һЩ��������Щ������ԭ���Ǹ�����������ϸ���ڶ������������������������ mRNA �� reads ������Ƚ��Ⱥ˻�����ı������ reads �ߣ����и�ˮƽ�Ļ����������Plese ��[99]������ Trimitomics �����ܿ�����Ч�ô�ת¼�� reads ���������������������މ��ƴװ���������ķ������̰����� NOVOPlasty�� Bowtie2/Trinity �� Velvet �� 3 ������ƴװ���̣�(1)����ʹ�� NOVOPlasty������ȫ���� RNA readsމ�д�ͷƴװ������ Kmer ��С��Χ(25��39��45 �� 51)ȷ��������������е�������;(2)���û��ƴװ��������������������л���ƴװ���������У�����ʹ�� Trimmomatic 0.33[100]��ԭʼ RNA readsމ�й��ˣ����� Bowtie2[64]���������˺�� reads �ȶԵ���Ե���ֵ�������������ϣ��� Trinity[94,101]�� mappedread މ�д�ͷƴװ;(3)ʹ�� Velvet ������ȫ����ת¼��މ�д�ͷƴװ�������� BlastN ����[102]ȷ���õ��������� Contigs��������� 3 �ַ�����û��ƴװ��������������������У���ô��ʹ�� Geneious ������������ 3 �ַ���ƴװ�Ľ�����ٽ����ϵĽ���� NCBI ���ݿ���މ��ͬԴ�Լ���������ͨ���� 6 ��������މ���������������ƴװ��������� 3 ��ƴװ���̶��ܹ����ǵ� 97%���ϵ����������������С���ƴװ�����Ժ�ȷ�������� NOVOPlasty�� Bowtie2/Trinity �� Velvet ƴװ���̵Ŀɿ��ԣ�������� 3 ��ƴװ���������ֲ�������죬�� A.valida �� P.dumerilii ������Ŧ�ζ��Bowtie2/Trinity ƴװ���̵õ���������������е��������á���������ʱ�䡢�����ڴ��ϱȽϣ�NOVOPlasty ƴװ���̸��������ơ�ֵ��ע����ǣ�Trimitomics �����ṩ 3 ��ƴװ���̣�ͨ���ж�ƴװ��������������ж��Ƿ�މ������ƴװ���̡�ͬʱ���ڸ������ֵ�����������飬���������� 3 ��ƴװ���̵Ľ���������˿ɿ��ԡ� ——�������ߣ�����������
SCISSCIAHCI